sylph is a program that performs ultrafast (1) ANI querying or (2) metagenomic profiling for metagenomic shotgun samples.
Containment ANI querying: sylph can search a genome, e.g. E. coli, against your sample. If sylph outputs an estimate of 97% ANI, your sample contains an E. coli with 97% ANI to the queried genome.
Metagenomic profiling: sylph can determine the species/taxa in your sample and their abundances, just like Kraken or MetaPhlAn.
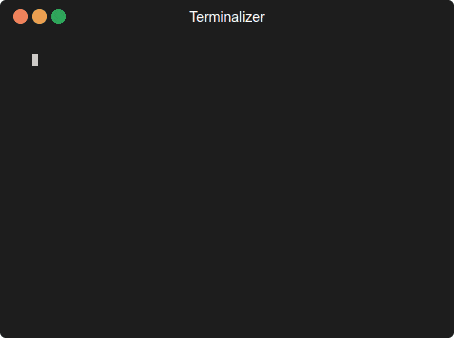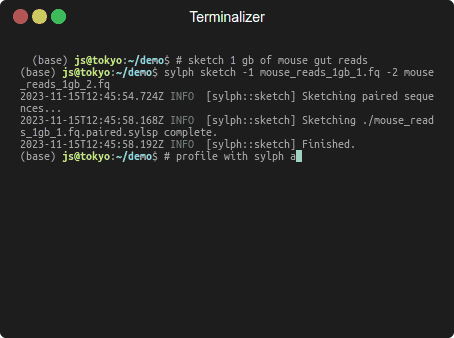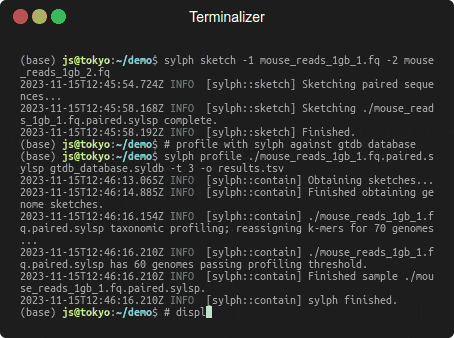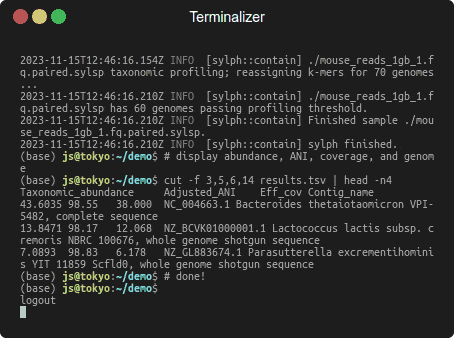
Profiling 1 Gbp of mouse gut reads against 85,205 genomes in a few seconds
-
Precise species-level profiling: sylph has less false positives than Kraken and is about as precise and sensitive as marker gene methods (MetaPhlAn, mOTUs).
-
Ultrafast, multithreaded, multi-sample: sylph can be > 50x faster than other methods. Sylph only takes ~15GB of RAM for profiling against the entire GTDB-R220 database (110k genomes).
-
Accurate (containment) ANI information: sylph can give accurate ANI estimates between reference genomes and your metagenome sample down to 0.1x coverage.
-
Customizable databases and pre-built databases: We offer pre-built databases of prokaryotes, viruses, eukaryotes. Custom databases (e.g. using your own MAGs) are easy to build.
-
Short or long reads: Sylph was also the most accurate method on Oxford Nanopore's independent benchmarks.
sylph uses a k-mer containment method. sylph's novelty lies in using a statistical technique to estimate k-mer containment for low coverage genomes , giving accurate results for low abundance organisms. See here for more information on what sylph can and can not do.
Profile metagenome sample against GTDB-R220 (113,104 bacterial/archaeal species representative genomes)
conda install -c bioconda sylph
# download GTDB-R220 pre-built database (~13 GB)
wget http://faust.compbio.cs.cmu.edu/sylph-stuff/gtdb-r220-c200-dbv1.syldb
# multi-sample paired-end profiling (sylph version >= 0.6)
sylph profile gtdb-r220-c200-dbv1.syldb -1 *_1.fastq.gz -2 *_2.fastq.gz -t (threads) > profiling.tsv
# multi-sample single-end profiling
sylph profile gtdb-r220-c200-dbv1.syldb *.fastq -t (threads) > profiling.tsv

conda install -c bioconda sylphRequirements:
- rust (version > 1.63) programming language and associated tools such as cargo are required and assumed to be in PATH.
- A c compiler (e.g. GCC)
- make
- cmake
Building takes a few minutes (depending on # of cores).
git clone https://github.com/bluenote-1577/sylph
cd sylph
# If default rust install directory is ~/.cargo
cargo install --path . --root ~/.cargo
sylph profile test_files/*If you're on an x86-64 system, you can download the binary and use it without any installation.
wget https://github.com/bluenote-1577/sylph/releases/download/latest/sylph
chmod +x sylph
./sylph -hNote: the binary is compiled with a different set of libraries (musl instead of glibc), probably impacting performance.
The pre-built databases available here can be downloaded and used with sylph for profiling and containment querying.
For common use cases and fast explanations, see the above cookbook.
To incorporate taxonomy into sylph's outputs, see the sylph-tax repository.
Tip
The new sylph-tax program replaces the old sylph-utils repository.
- Made the
inspectoption much less memory intensive. Slightly changed outputs when no genomes are found.
See the CHANGELOG for complete details.
Jim Shaw and Yun William Yu. Rapid species-level metagenome profiling and containment estimation with sylph (2024). Nature Biotechnology.