This is a repository that I use for this course. My assignments are submitted here.
write_razor_pbs.py: This python script generates an output which can be saved as a bash (.sh) script for submitting jobs to the Razor cluster on the Uark AHPCC using PBS scheduling syntax.write_pinnacle_slurm.py: This python script generates an output which can be saved as a bash (.sh) script for submitting jobs to the Pinnacle cluster on the Uark AHPCC using SLURM scheduling syntax.nucleotide_composition.py: This python script calculates the frequency of nucleotides of a string of characters of a nucleotide sequence (requires removal of header prior to entering into script, as written). Created in completion of Assignment 5 of this course, not written to be practical for everyday use as is.parseGFF.py: This python script runs from command line, takes two inputs (1) a .gff file with genomic features and (2) a .fasta file of the corresponding genome. This script extracts the sequence of each CDS feature of the gff file, combines exons in the correct order, and returns whole exons as individual fasta formatted sequences to STDOUT. The reverse complement sequence will be used for features that are coded to be on the "-" strand in the input gff file.
CV.md: This file is my curriculum vitae written in markdown format.assn03.tgz: The assn03.tgz file is a zipped tape archive containing the bash scripts that I used to create BLAST databases and complete BLAST searches, as well as their outputs and answers to all questions in completion of assignment 3 questions in this course.Supplemental_files: A folder that contains README images, and other "clutter" in this repo.Assn_7_help.txt: A file with notes on how to check if the parseGFF.py file is working correctly. How to compare parseGFF.py output with provided fasta sequences for these genes.
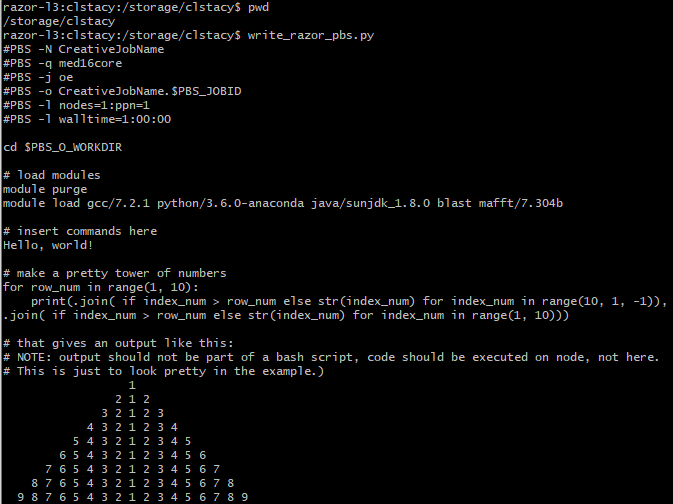
Here is an example output of python script that generates PBS bash scripts for submission on the razor cluster:
 ]
]