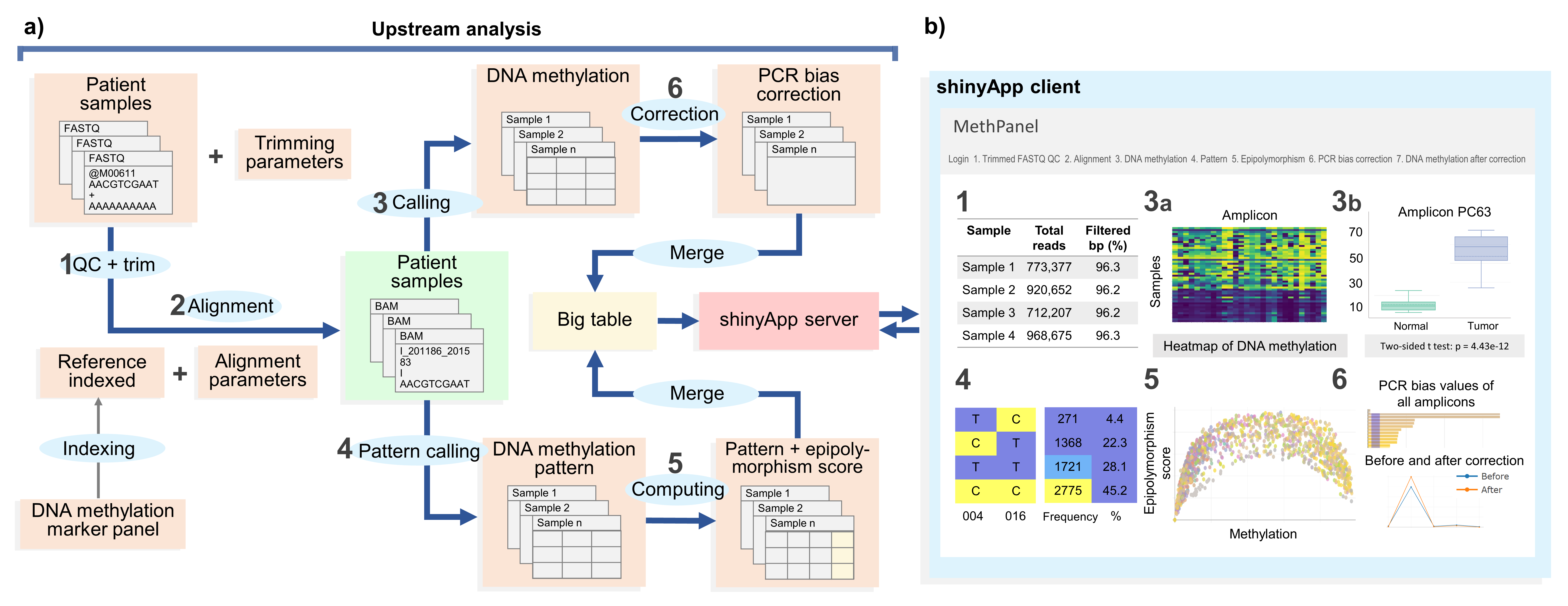
MethPanel is a computational pipeline in Linux operating system with an interactive graphical interface for rapid analysis of multiplex bisulphite PCR sequencing data. The tool covers a complete analysis workflow from genomic alignment to DNA methylation calling and supports an unlimited number of PCR amplicons and input samples. Moreover MethPanel offers important and unique features, such as a epipolymorphism score and a bisulphite PCR bias correction. MethPanel can be run in parallel by samples on either a personal computer or a high performance computer. The outputs are automatically forwarded to a shinyApp for convenient display, visualisation and sharing of data with collaborators and clinicians.
Before applying our MethPanel workflow, all the primers for the multiplex bisulphite PCR can be designed using online tool PrimerSuite (Lu J, Johnston A, Berichon P, Ru K-l, Korbie D, Trau M: PrimerSuite: a high throughput web-based primer design program for multiplex bisulphite PCR. Scientific reports 2017, 7:41328).
-
MethPanel is built based on in-house bash/python/R script, Bpipe and a collection of software packages:
- [bowtie2, 2.3.5.1] http://bowtie-bio.sourceforge.net/bowtie2/index.shtml
- [Bismark, 0.22.3] https://www.bioinformatics.babraham.ac.uk/projects/bismark/
- [bedtools, 2.28.0] https://github.com/arq5x/bedtools2
- [samtools, 1.9] https://github.com/samtools/
- [Bpipe, 0.9.9.9] https://github.com/ssadedin/bpipe
- [fastqc, 0.11.9] http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- [trim_galore, 0.6.5] https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- [python, 3.7.4] https://www.python.org/
- [python modules] configobj, argparse
- [R, 3.6.1] https://www.r-project.org/
- [R and Bioconductor packages] rtracklayer, Biostrings, GenomicRanges, data.table, ggplot2, gplots, reshape2, shiny, shinyauthr, shinyjs, plotly
- [multiqc] https://multiqc.info/
- [java, jdk-13.33] https://java.com/en/download/
- [shiny server] Visit our Wiki page for a detailed installation manual of shiny server and shinyapp
-
Dependencies
Download this script to automatically download all the above listed software packages, install python and R packages, and clone MethPanel github repository. After downloading, execute the command below:
bash ./install_dependencies.shOr manually download the above listed software packages, install python and R packages
-
[python version >= 3.7.4]
python -m pip install configobj argparse -
[R version >= 3.6.1]
install.packages(c("ggplot2", "data.table", "gplots", "reshape2", "shiny", "shinyauthr", "shinyjs", "plotly"))if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager") BiocManager::install(c("Biostrings", "GenomicRanges", "rtracklayer")) -
Clone MethPanel github repository
git clone git@github.com:/thinhong/MethPanel.git- or
git clone https://github.com/thinhong/MethPanel.git
-
-
Inputs required
- Single/paired fastq files in .gz format for each sample
- Filled sample config file, example
- Filled system config file, example
- DNA methylation marker panel file, example
- Note:
- The fastq files are located in
/path/to/upstream/analysis/<user>/${project}/raw/Sample1/Sample1_R1.fastq.gz,/path/to/upstream/analysis/<user>/${project}/raw/Sample1/Sample1_R2.fastq.gz(if paired-end) for each sample.
- The fastq files are located in
project="<project_name>"
sample_config="/path/to/upstream/analysis/<user>/${project}/config/sample.${project}.pre.config"
system_config="/path/to/upstream/analysis/<user>/${project}/config/system.${project}.pre.config"
The fastq files are located in /path/to/upstream/analysis/<user>/<project_name>/raw/
python "/path/to/pipe/run_Bpipe.py" $sample_config $system_config
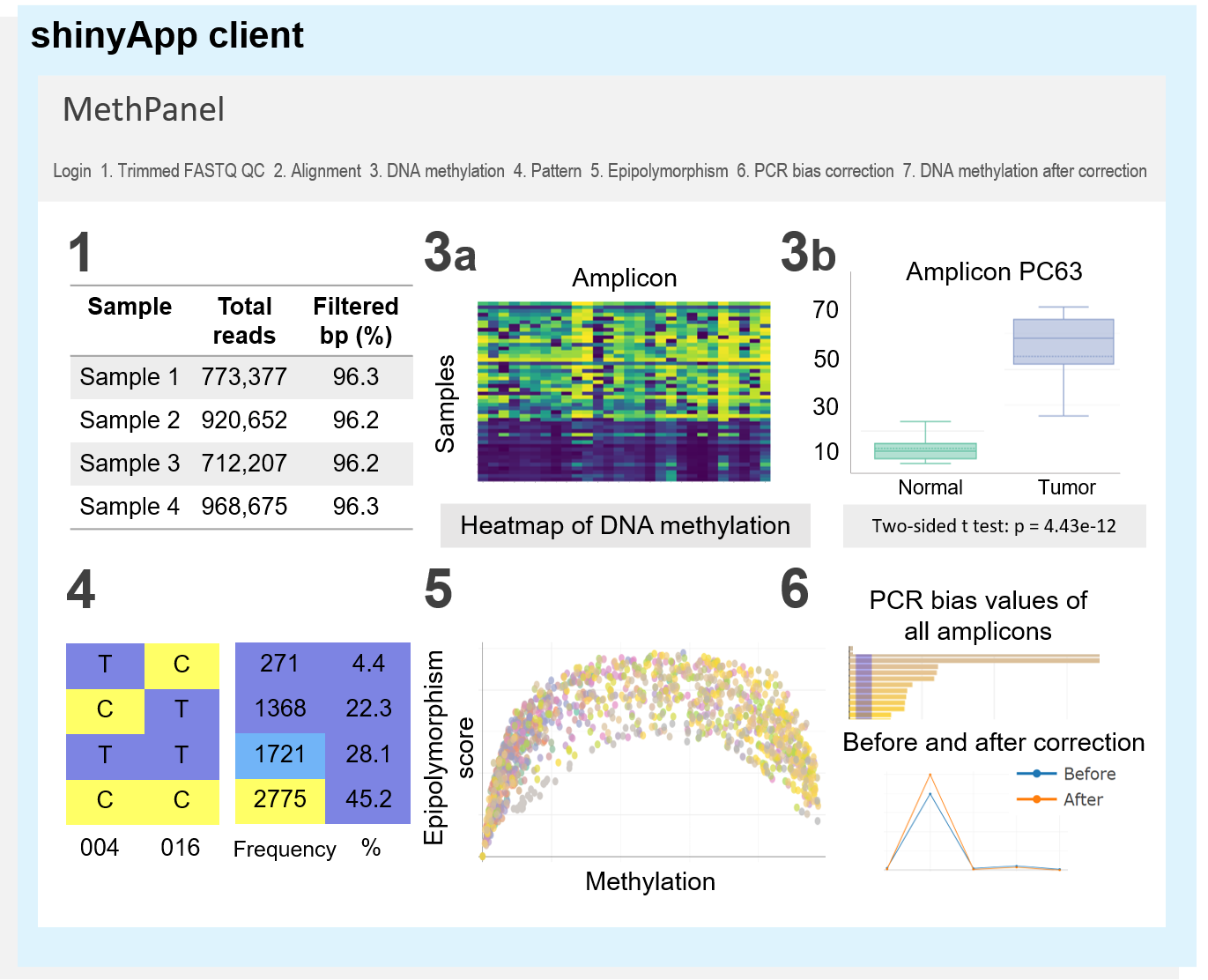
For further details manual of MethPanel shinyApp, please visit our Wiki page.