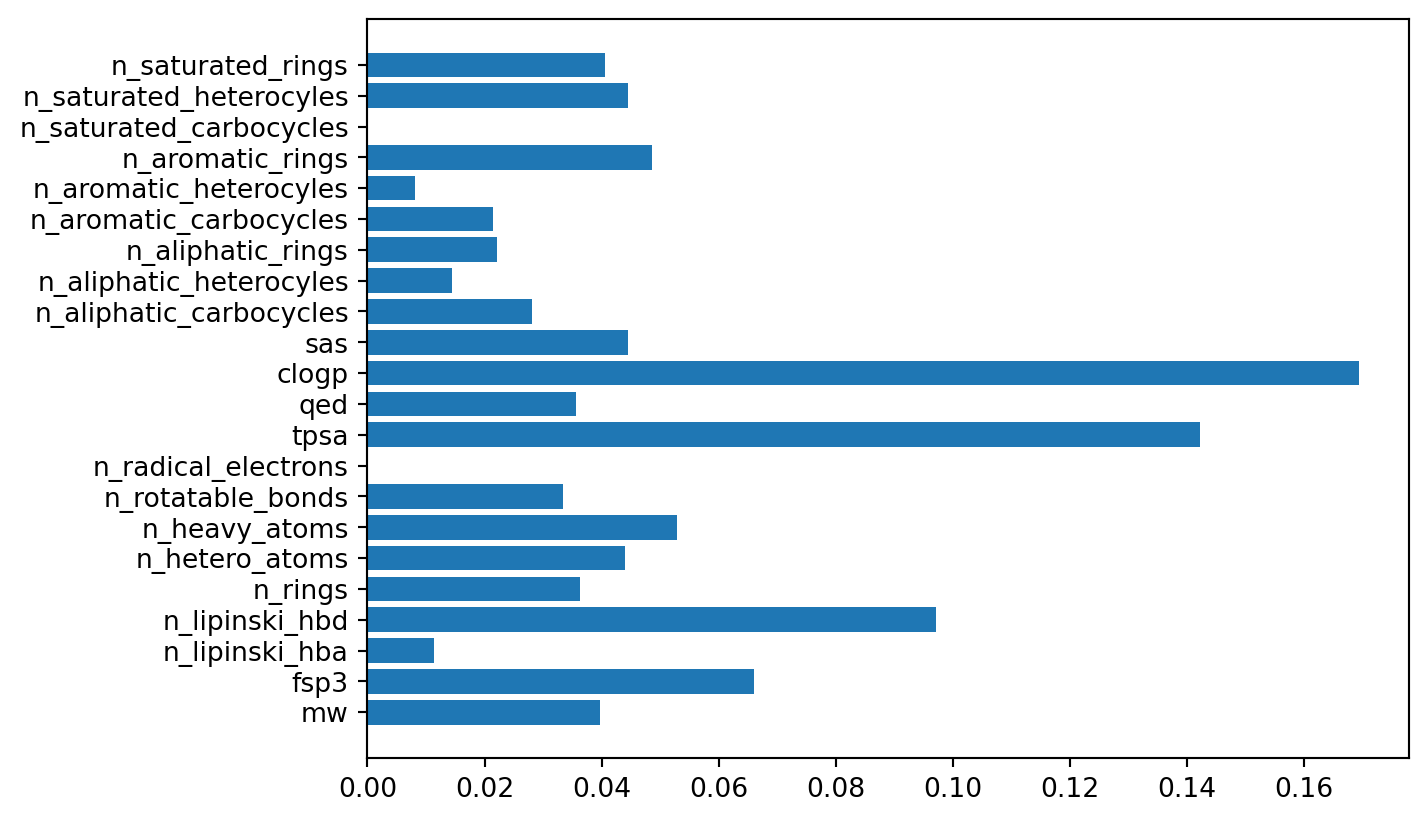
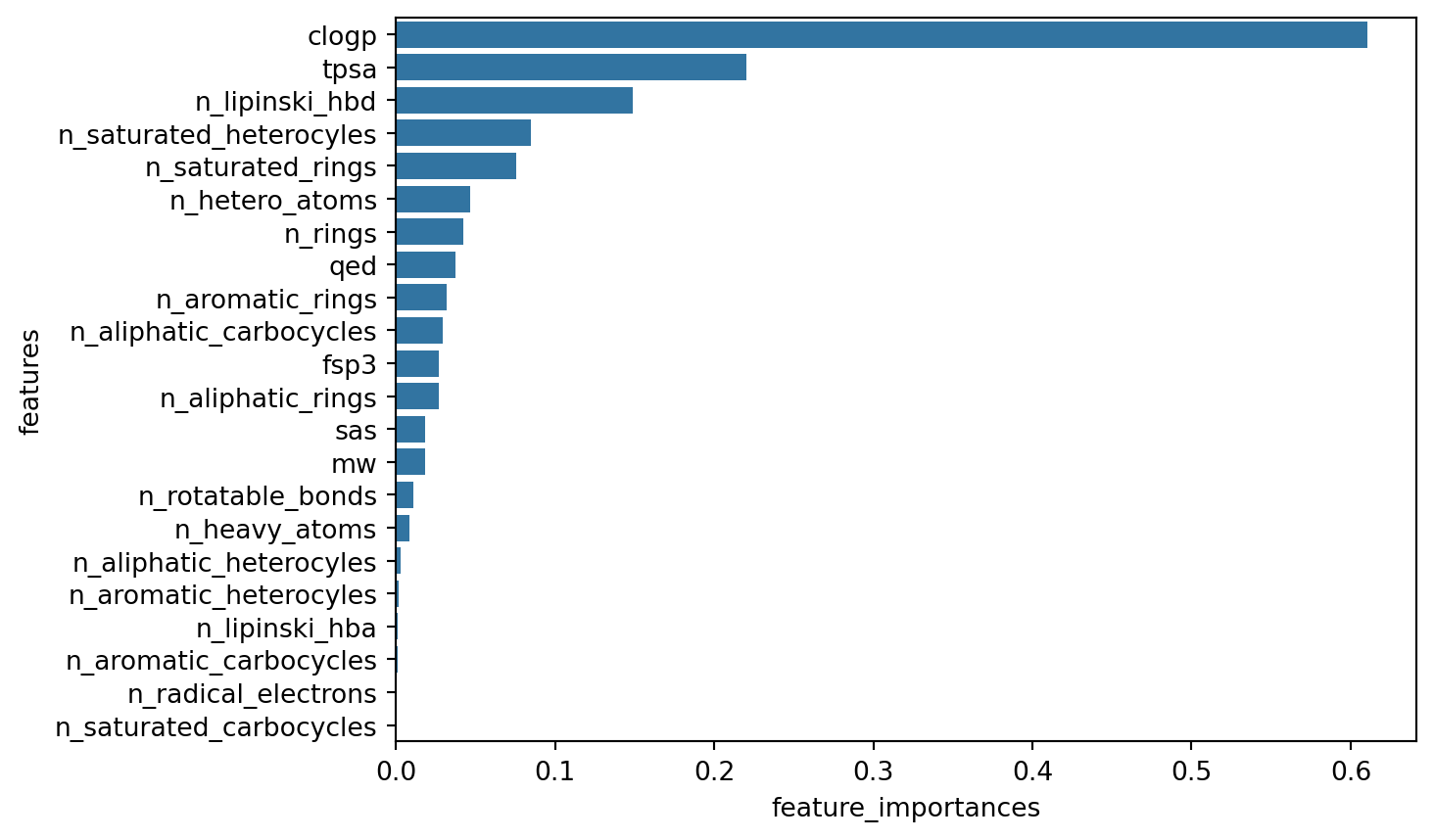
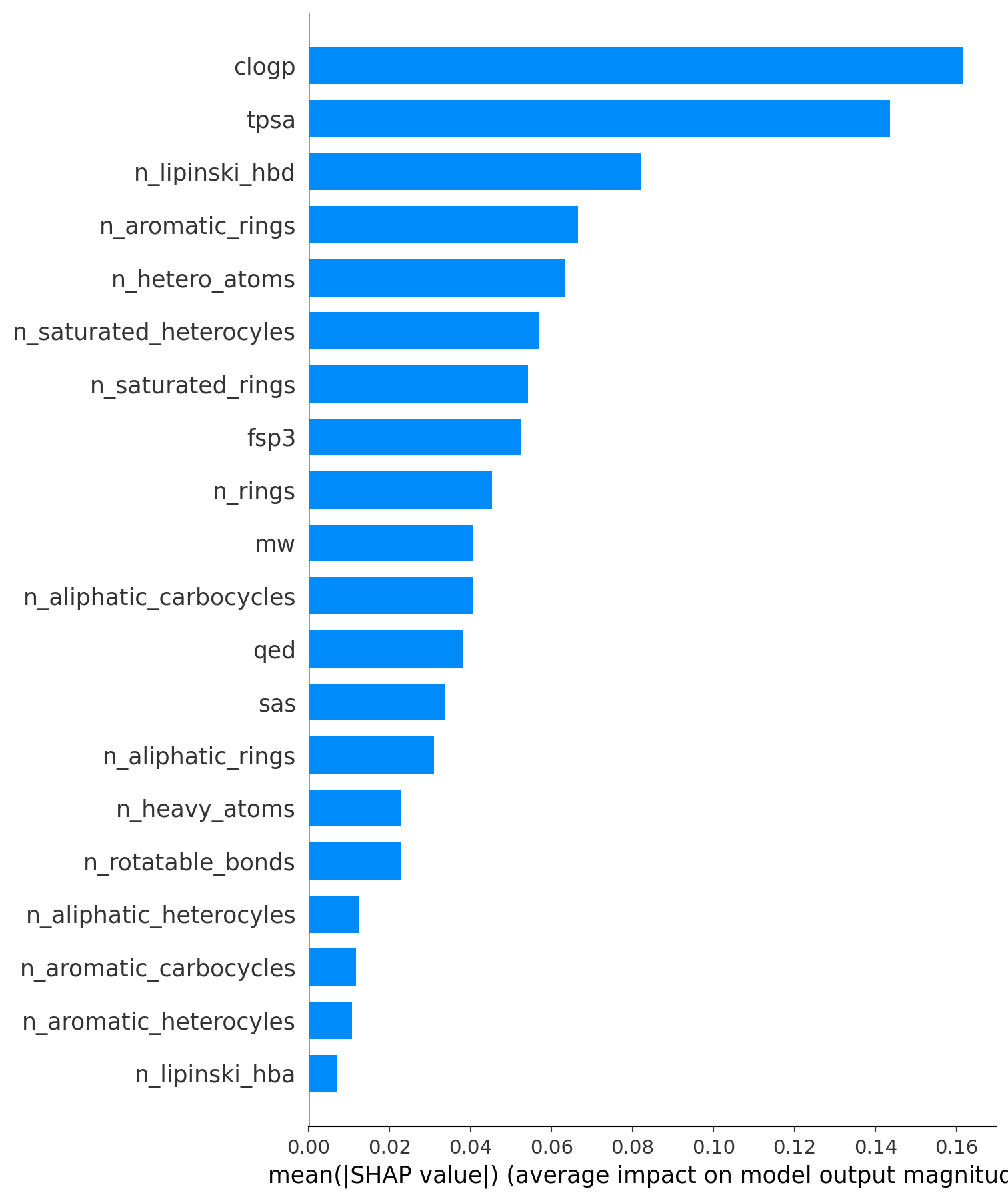
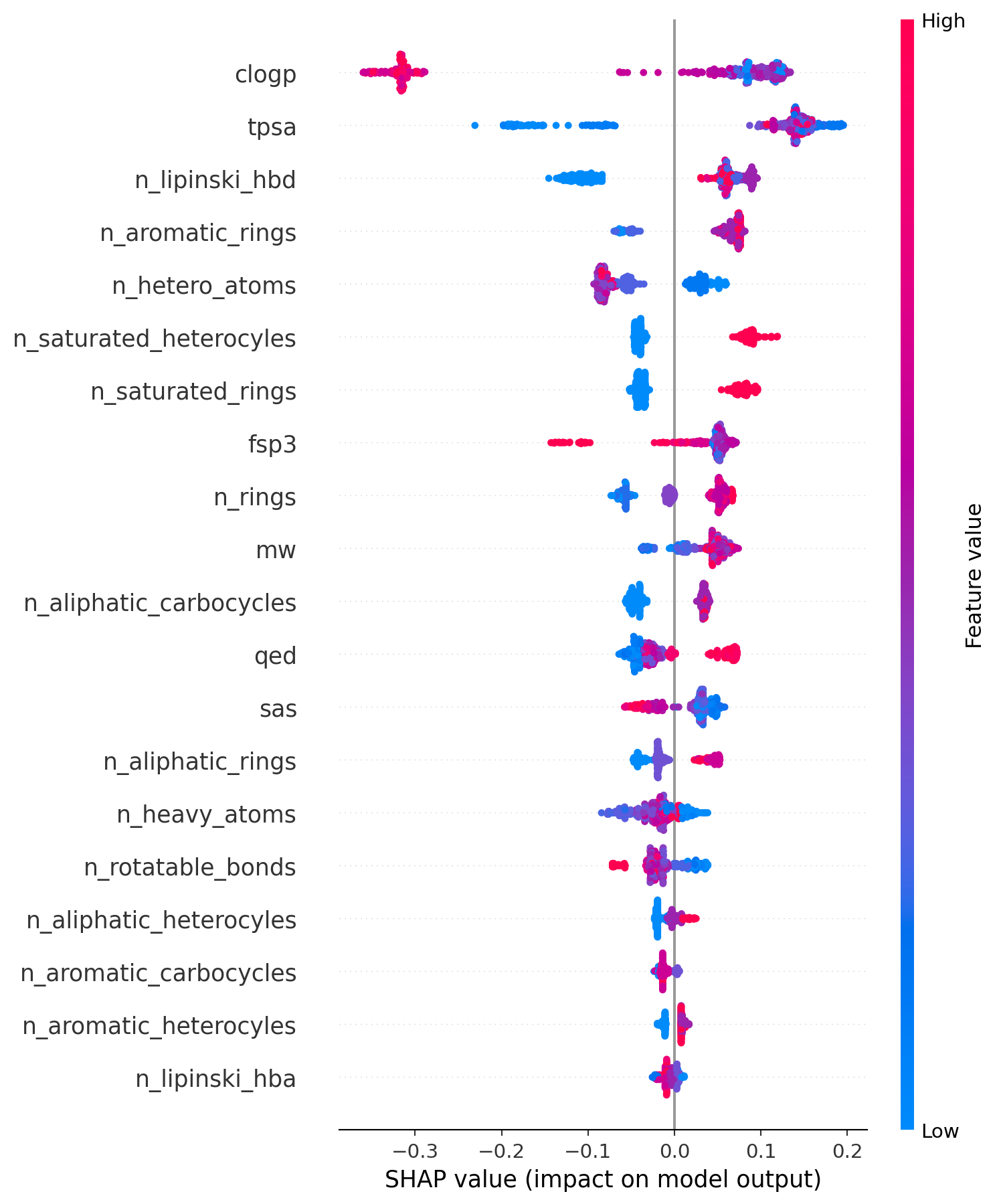
diff --git a/.nojekyll b/.nojekyll
index 9853f45..ed88895 100644
--- a/.nojekyll
+++ b/.nojekyll
@@ -1 +1 @@
-31110224
\ No newline at end of file
+53b3dcd9
\ No newline at end of file
diff --git a/index-python.xml b/index-python.xml
index 1b3d98b..54d65dc 100644
--- a/index-python.xml
+++ b/index-python.xml
@@ -10,7 +10,1230 @@
A blog website about using software tools to decipher drug discovery and development dataquarto-1.2.280
-Wed, 21 Aug 2024 12:00:00 GMT
+Tue, 07 Jan 2025 11:00:00 GMT
+
+ Building a simple deep learning model about adverse drug reactions
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/22_Simple_dnn_adrs/2_ADR_regressor.html
+ The notebook from this repository uses a venv created by using uv with a kernel set up this way.
+
Some of the code blocks have been folded to keep the post length a bit more manageable - click on the code links to see full code (only applies to the HTML version, not the Jupyter notebook version).
+
+
+
Import libraries
+
+
+Code
+
import pandas as pd
+import torch
+import torch.nn as nn
+import torch.nn.functional as F
+from torch.nn.functional import one_hot
+from torch.utils.data import TensorDataset, DataLoader
+import numpy as np
+import datamol as dm
+import rdkit
+from rdkit import Chem
+from rdkit.Chem import rdFingerprintGenerator
+import useful_rdkit_utils as uru
+import sys
+from matplotlib import pyplot as plt
+print(f"Pandas version used: {pd.__version__}")
+print(f"PyTorch version used: {torch.__version__}")
+print(f"NumPy version used: {np.__version__}")
+print(f"RDKit version used: {rdkit.__version__}")
+print(f"Python version used: {sys.version}")
+
+
+
Pandas version used: 2.2.3
+PyTorch version used: 2.2.2
+NumPy version used: 1.26.4
+RDKit version used: 2024.09.4
+Python version used: 3.12.7 (v3.12.7:0b05ead877f, Sep 30 2024, 23:18:00) [Clang 13.0.0 (clang-1300.0.29.30)]
+
+
+
+
+
+
Import adverse drug reactions (ADRs) data
+
This is an extremely small set of data compiled manually (by me) via references stated in the dataframe. For details about what and how the data are collected, I’ve prepared a separate post as a data note (add post link) to explain key things about the data. It may not lead to a very significant result but it is done as an example of what an early or basic deep neural network (DNN) model may look like. Ideally there should be more training data and also more features added or used. I’ve hypothetically set the goal of this introductory piece to predict therapeutic drug classes from ADRs, molecular fingerprints and cytochrome P450 substrate strengths, but this won’t be achieved in this initial post (yet).
+
+
data = pd.read_csv("All_CYP3A4_substrates")
+print(data.shape)
+data.head(3)
+
+
(27, 8)
+
+
+
+
+
+
+
+
+
+
generic_drug_name
+
notes
+
cyp_strength_of_evidence
+
drug_class
+
adverse_drug_reactions
+
first_ref
+
second_ref
+
date_checked
+
+
+
+
+
0
+
carbamazepine
+
NaN
+
strong
+
antiepileptics
+
constipation^^, leucopenia^^, dizziness^^, som...
+
drugs.com
+
nzf
+
211024
+
+
+
1
+
eliglustat
+
NaN
+
strong
+
metabolic_agents
+
diarrhea^^, oropharyngeal_pain^^, arthralgia^^...
+
drugs.com
+
emc
+
151124
+
+
+
2
+
flibanserin
+
NaN
+
strong
+
CNS_agents
+
dizziness^^, somnolence^^, sedation^, fatigue^...
+
drugs.com
+
Drugs@FDA
+
161124
+
+
+
+
+
+
+
For drug with astericks marked in “notes” column, see data notes under “Exceptions for ADRs” section in 1_ADR_data.qmd (separate post).
+
I’m dropping some of the columns that are not going to be used later.
Before extracting data from ChEMBL, I’m getting a list of drug names in capital letters ready first which can be fed into chembl_downloader with my old cyp_drugs.py to retrieve the SMILES of these drugs.
+
+
+Code
+
string = df["generic_drug_name"].tolist()
+# Convert list of drugs into multiple strings of drug names
+drugs =f"'{"','".join(string)}'"
+# Convert from lower case to upper case
+for letter in drugs:
+if letter.islower():
+ drugs = drugs.replace(letter, letter.upper())
+print(drugs)
# Get SMILES for each drug (via copying-and-pasting the previous cell output - attempted various ways to feed the string
+# directly into cyp_drugs.py, current way seems to be the most straightforward one...)
+from cyp_drugs import chembl_drugs
+# Using ChEMBL version 34
+df_3a4 = chembl_drugs(
+'CARBAMAZEPINE','ELIGLUSTAT','FLIBANSERIN','IMATINIB','IBRUTINIB','NERATINIB','ESOMEPRAZOLE','OMEPRAZOLE','IVACAFTOR','NALOXEGOL','OXYCODONE','SIROLIMUS','TERFENADINE','DIAZEPAM','HYDROCORTISONE','LANSOPRAZOLE','PANTOPRAZOLE','LERCANIDIPINE','NALDEMEDINE','NELFINAVIR','TELAPREVIR','ONDANSETRON','QUININE','RIBOCICLIB','SUVOREXANT','TELITHROMYCIN','TEMSIROLIMUS',
+#file_name="All_cyp3a4_smiles"
+ )
+print(df_3a4.shape)
+df_3a4.head(3)
+
+## Note: latest ChEMBL version 35 (as from 1st Dec 2024) seems to be taking a long time to load (no output after ~7min),
+## both versions 33 & 34 are ok with outputs loading within a few secs
+
+
+
(27, 4)
+
+
+
+
+
+
+
+
+
+
chembl_id
+
pref_name
+
max_phase
+
canonical_smiles
+
+
+
+
+
0
+
CHEMBL108
+
CARBAMAZEPINE
+
4
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
+
+
1
+
CHEMBL12
+
DIAZEPAM
+
4
+
CN1C(=O)CN=C(c2ccccc2)c2cc(Cl)ccc21
+
+
+
2
+
CHEMBL2110588
+
ELIGLUSTAT
+
4
+
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...
+
+
+
+
+
+
+
+
+
+
Merge dataframes
+
Next, I’m renaming the drug name column and merging the two dataframes together where one contains the ADRs and the other one contains the SMILES. I’m also making sure all drug names are in upper case for both dataframes so they can merge properly.
Then I’m parsing the canonical SMILES through my old script to generate these small molecules as RDKit molecules and standardised SMILES, making sure these SMILES are parsable.
+
+
+Code
+
# Using my previous code to preprocess small mols
+# disable rdkit messages
+dm.disable_rdkit_log()
+
+# The following function code were adapted from datamol.io
+def preprocess(row):
+
+"""
+ Function to preprocess, fix, standardise, sanitise compounds
+ and then generate various molecular representations based on these molecules.
+ Can be utilised as df.apply(preprocess, axis=1).
+
+ :param smiles_column: SMILES column name (needs to be names as "canonical_smiles")
+ derived from ChEMBL database (or any other sources) via an input dataframe
+ :param mol: RDKit molecules
+ :return: preprocessed RDKit molecules, standardised SMILES, SELFIES,
+ InChI and InChI keys added as separate columns in the dataframe
+ """
+
+# smiles_column = strings object
+ smiles_column ="canonical_smiles"
+# Convert each compound into a RDKit molecule in the smiles column
+ mol = dm.to_mol(row[smiles_column], ordered=True)
+# Fix common errors in the molecules
+ mol = dm.fix_mol(mol)
+# Sanitise the molecules
+ mol = dm.sanitize_mol(mol, sanifix=True, charge_neutral=False)
+# Standardise the molecules
+ mol = dm.standardize_mol(
+ mol,
+# Switch on to disconnect metal ions
+ disconnect_metals=True,
+ normalize=True,
+ reionize=True,
+# Switch on "uncharge" to neutralise charges
+ uncharge=True,
+# Taking care of stereochemistries of compounds
+# Note: this uses the older approach of "AssignStereochemistry()" from RDKit
+# https://github.com/datamol-io/datamol/blob/main/datamol/mol.py#L488
+ stereo=True,
+ )
+
+# Adding following rows of different molecular representations
+ row["rdkit_mol"] = dm.to_mol(mol)
+ row["standard_smiles"] = dm.standardize_smiles(str(dm.to_smiles(mol)))
+#row["selfies"] = dm.to_selfies(mol)
+#row["inchi"] = dm.to_inchi(mol)
+#row["inchikey"] = dm.to_inchikey(mol)
+return row
+
+df_p3a4 = df.apply(preprocess, axis =1)
+print(df_p3a4.shape)
+df_p3a4.head(3)
+
+
+
(27, 9)
+
+
+
+
+
+
+
+
+
+
pref_name
+
cyp_strength_of_evidence
+
drug_class
+
adverse_drug_reactions
+
chembl_id
+
max_phase
+
canonical_smiles
+
rdkit_mol
+
standard_smiles
+
+
+
+
+
0
+
CARBAMAZEPINE
+
strong
+
antiepileptics
+
constipation^^, leucopenia^^, dizziness^^, som...
+
CHEMBL108
+
4
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2dee0>
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
+
+
1
+
ELIGLUSTAT
+
strong
+
metabolic_agents
+
diarrhea^^, oropharyngeal_pain^^, arthralgia^^...
+
CHEMBL2110588
+
4
+
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2dfc0>
+
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...
+
+
+
2
+
FLIBANSERIN
+
strong
+
CNS_agents
+
dizziness^^, somnolence^^, sedation^, fatigue^...
+
CHEMBL231068
+
4
+
O=c1[nH]c2ccccc2n1CCN1CCN(c2cccc(C(F)(F)F)c2)CC1
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2e030>
+
O=c1[nH]c2ccccc2n1CCN1CCN(c2cccc(C(F)(F)F)c2)CC1
+
+
+
+
+
+
+
+
+
+
Split data
+
Random splits usually lead to overly optimistic models, where testing molecules are too similar to traininig molecules leading to many problems. This is further discussed in two other blog posts that I’ve found useful - post by Greg Landrum and post by Pat Walters.
+
Here I’m trying out Pat’s useful_rdkit_utils’ GroupKFoldShuffle code (code originated from this thread) to split data (Butina clustering/splits). To do this, it requires SMILES to generate molecular fingerprints which will be used in the training and testing sets (potentially for future posts and in real-life cases, more things can be done with the SMILES or other molecular representations for machine learning, but to keep this post easy-to-read, I’ll stick with only generating the Morgan fingerprints for now).
+
+
+Code
+
# Generate numpy arrays containing the fingerprints
+df_p3a4['fp'] = df_p3a4.rdkit_mol.apply(rdFingerprintGenerator.GetMorganGenerator().GetCountFingerprintAsNumPy)
+
+# Get Butina cluster labels
+df_p3a4["butina_cluster"] = uru.get_butina_clusters(df_p3a4.standard_smiles)
+
+# Set up a GroupKFoldShuffle object
+group_kfold_shuffle = uru.GroupKFoldShuffle(n_splits=5, shuffle=True)
+
+# Using cross-validation/doing data split
+## X = np.stack(df_s3a4.fp), y = df.adverse_drug_reactions, group labels = df_s3a4.butina_cluster
+for train, test in group_kfold_shuffle.split(np.stack(df_p3a4.fp), df.adverse_drug_reactions, df_p3a4.butina_cluster):
+print(len(train),len(test))
+
+
+
17 10
+23 4
+23 4
+23 4
+22 5
+
+
+
+
+
+
Locate training and testing sets after data split
+
While trying to figure out how to locate training and testing sets after the data split, I’ve gone into a mini rabbit hole myself (a self-confusing session but gladly it clears up when my thought process goes further…). For example, some of the ways I’ve planned to try: create a dictionary as {index: butina label} first - butina cluster labels vs. index e.g. df_s3a4[“butina_cluster”], or maybe can directly convert from NumPy array to tensor - will need to locate drugs via indices first to specify training and testing sets, e.g. torch_train = torch.from_numpy(train) or torch_test = torch.from_numpy(test). It is actually simpler than this, which is to use pd.DataFrame.iloc() as shown below.
# What df_p3a4 now looks like after data split - with "fp" and "butina_cluster" columns added
+df_p3a4.head(1)
+
+
+
+
+
+
+
+
+
pref_name
+
cyp_strength_of_evidence
+
drug_class
+
adverse_drug_reactions
+
chembl_id
+
max_phase
+
canonical_smiles
+
rdkit_mol
+
standard_smiles
+
fp
+
butina_cluster
+
+
+
+
+
0
+
CARBAMAZEPINE
+
strong
+
antiepileptics
+
constipation^^, leucopenia^^, dizziness^^, som...
+
CHEMBL108
+
4
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2dee0>
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ...
+
20
+
+
+
+
+
+
+
+
# Convert indices into list
+train_set = train.tolist()
+# Locate drugs and drug info via pd.DataFrame.iloc
+df_train = df_p3a4.iloc[train_set]
+print(df_train.shape)
+df_train.head(2)
Set up training and testing sets for X and y variables
+
This part involves converting X (features) and y (target) variables into either one-hot encodings or vector embeddings, since I’ll be dealing with categories/words/ADRs and not numbers, and also to split each X and y variables into training and testing sets. At the very beginning, I’ve thought about using scikit_learn’s train_test_split(), but then realised that I should not need to do this as it’s already been done in the previous step (obviously I’m confusing myself again…). Essentially, this step can be integrated with the one-hot encoding and vector embeddings part as shown below.
+
There are three coding issues that have triggered warning messages when I’m trying to figure out how to convert CYP strengths into one-hot encodings:
+
+
A useful thread has helped me to solve the downcasting issue in pd.DataFrame.replace() when trying to do one-hot encoding to replace the CYP strengths for each drug
+
A Pandas setting-with-copy warning shows if using df[“column_name”]:
+
+
+
A value is trying to be set on a copy of a slice from a DataFrame. Try using .loc[row_indexer,col_indexer] = value instead
+
+
The solution is to enable the copy-on-write globally (as commented in the code below; from Pandas reference).
+
+
PyTorch user warning appers if using df_train[“cyp_strength_of_evidence”].values, as this leads to non-writable tensors with a warning like this:
+
+
+
UserWarning: The given NumPy array is not writable, and PyTorch does not support non-writable tensors. This means writing to this tensor will result in undefined behavior. You may want to copy the array to protect its data or make it writable before converting it to a tensor. This type of warning will be suppressed for the rest of this program. (Triggered internally at /Users/runner/work/pytorch/pytorch/pytorch/torch/csrc/utils/tensor_numpy.cpp:212.)
+
+
One of the solutions is to add copy() e.g. col_encoded = one_hot(torch.from_numpy(df[“column_name”].values.copy()) % total_numbers_in_column) or alternatively, convert column into numpy array first, then make the numpy array writeable (which is what I’ve used in the code below).
+
+
+Code
+
## X_train
+# 1. Convert "cyp_strength_of_evidence" column into one-hot encoding
+# Enable copy-on-write globally to remove the warning
+pd.options.mode.copy_on_write =True
+
+# Replace CYP strength as numbers
+with pd.option_context('future.no_silent_downcasting', True):
+ df_train["cyp_strength_of_evidence"] = df_train["cyp_strength_of_evidence"].replace({"strong": 1, "mod": 2}).infer_objects()
+ df_test["cyp_strength_of_evidence"] = df_test["cyp_strength_of_evidence"].replace({"strong": 1, "mod": 2}).infer_objects()
+
+# Get total number of CYP strengths in df
+total_cyp_str_train =len(set(df_train["cyp_strength_of_evidence"]))
+
+# Convert column into numpy array first, then make the numpy array writeable
+cyp_array_train = df_train["cyp_strength_of_evidence"].to_numpy()
+cyp_array_train.flags.writeable =True
+cyp_str_train_t = one_hot(torch.from_numpy(cyp_array_train) % total_cyp_str_train)
+cyp_str_train_t
Without going into too much details about vector embeddings (as there are a lot of useful learning materials about it online and in texts), here’s roughly how I understand embeddings while working on this post. Embeddings are real-valued dense vectors that are normally in multi-dimensional arrays and they can represent and catch the context of a word or sentence, the semantic similarity and especially the relation of each word with other words in a corpus of texts. They roughly form the basis of natural language processing and also contribute to how large language models are built… in a very simplified sense, but obviously this can get complex if we want the models to do more. Here, I’m trying something experimental so I’m going to convert each ADR for each drug into embeddings.
+
+
+Code
+
# 2. Convert "adverse_drug_reactions" column into embeddings
+## see separate scripts used previously e.g. words_tensors.py
+## or Tensors_for_adrs_interactive.py to show step-by-step conversions from words to tensors
+
+# Save all ADRs from common ADRs column as a list (joining every row of ADRs in place only)
+adr_str_train = df_train["adverse_drug_reactions"].tolist()
+# Join separate rows of strings into one complete string
+adr_string_train =",".join(adr_str_train)
+# Converting all ADRs into Torch tensors using words_tensors.py
+from words_tensors import words_tensors
+adr_train_t = words_tensors(adr_string_train)
+adr_train_t
When trying to convert the “fp” column into tensors, there is one coding issue I’ve found relating to the data split step earlier. Each time the notebook is re-run with the kernel refreshed, the data split will lead to different proportions of training and testing sets due to the “shuffle = True”, which subsequently leads to different training and testing set arrays. One of the ways to circumvent this is to turn off the shuffle but this is not ideal for model training. So an alternative way that I’ve tried is to use ndarray.size (which is the product of elements in ndarray.shape, equivalent to multiplying the numbers of rows and columns), and divide the row of the intended tensor shape by 2 as I’m trying to reshape training arrays so they’re all in 2 columns in order for torch.cat() to work later.
+
+
+Code
+
# 3. Convert "fp" column into tensors
+# Stack numpy arrays in fingerprint column
+fp_train_array = np.stack(df_train["fp"])
+# Convert numpy array data type from uint32 to int32
+fp_train_array = fp_train_array.astype("int32")
+# Create tensors from array
+fp_train_t = torch.from_numpy(fp_train_array)
+# Reshape tensors
+fp_train_t = torch.reshape(fp_train_t, (int(fp_train_array.size/2), 2))
+fp_train_t.shape # tensor.ndim to check tensor dimensions
+
+
+
torch.Size([22528, 2])
+
+
+
+
adr_train_t.shape
+
+
torch.Size([674, 2])
+
+
+
+
cyp_str_train_t.shape
+
+
torch.Size([22, 2])
+
+
+
+
# Concatenate adr tensors, fingerprint tensors and cyp strength tensors as X_train
+X_train = torch.cat([adr_train_t, fp_train_t, cyp_str_train_t], 0).float()
+X_train
## y_train
+# Use drug_class column as target
+# Convert "drug_class" column into embeddings
+# total number of drug classes in df = 20 - len(set(df["drug_class"])) - using embeddings instead of one-hot
+dc_str_train = df_train["drug_class"].tolist()
+dc_string_train =",".join(dc_str_train)
+y_train = words_tensors(dc_string_train)
+y_train
Input preprocessing pipeline using PyTorch Dataset and DataLoader
+
There is a size-mismatch-between-tensors warning when I’m trying to use PyTorch’s TensorDataset(). I’ve found out that to use the data loader and tensor dataset, the first dimension of all tensors needs to be the same. Initially, they’re not, where X_train.shape = [24313, 2], y_train.shape = [1, 2]. Eventually I’ve settled on two ways that can help with this:
+
+
use tensor.unsqueeze(dim = 1) or
+
use tensor[None] which’ll insert a new dimension at the beginning, then it becomes: X_train.shape = [1, 24313, 2], y_train.shape = [1, 1, 2]
+
+
+
X_train[None].shape
+
+
torch.Size([1, 23224, 2])
+
+
+
+
X_train.shape
+
+
torch.Size([23224, 2])
+
+
+
+
y_train[None].shape
+
+
torch.Size([1, 1, 2])
+
+
+
+
y_train.shape
+
+
torch.Size([1, 2])
+
+
+
+
# Create a PyTorch dataset on training data set
+train_data = TensorDataset(X_train[None], y_train[None])
+# Sets a seed number to generate random numbers
+torch.manual_seed(1)
+batch_size =1
+
+# Create a dataset loader
+train_dl = DataLoader(train_data, batch_size, shuffle =True)
+
+
+
# Create another PyTorch dataset on testing data set
+test_data = TensorDataset(X_test[None], y_test[None])
+torch.manual_seed(1)
+batch_size =1
+test_dl = DataLoader(test_data, batch_size, shuffle=True)
+
+
+
+
+
Set up a simple DNN regression model
+
I’m only going to use a very simple two-layer DNN model to match the tiny dataset used here. There are many other types of neural network layers or bits and pieces that can be used to suit the goals and purposes of the dataset used. This reference link shows different types of neural network layers that can be used in PyTorch.
+
Below are some short notes regarding a neural network (NN) model:
+
+
goal of the model is to minimise loss function L(W) (where W = weight) to get the optimal model weights
+
matrix with W (for hidden layer) connects input to hidden layer; matrix with W (for outer layer) connects hidden to output layer
+
Input layer -> activation function of hidden layer -> hidden layer -> activation function of output layer -> output layer (a very-simplified flow diagram to show how the layers get connected to each other)
+
+
About backpropagation for loss function:
+
+
backpropagation is a computationally efficient way to calculate partial derivatives of loss function to update weights in multi-layer NNs
+
it’s based on calculus chain rule to compute derivatives of mathematical functions (automatic differentiation)
+
matrix-vector multiplications in backpropagation are computationally more efficient to calculate than matrix-matrix multiplications e.g. forward propagation
# note: this is a very simple two-layer NN model only
+
+# Set up hidden units between two connected layers - one layer with 6 hidden units and the other with 3 hidden units
+hidden_units = [6, 3]
+# Input size same as number of columns in X_train
+input_size = X_train.shape[1]
+# Initiate NN layers as a list
+all_layers = []
+
+## Specify how the input, hidden and output layers are going to be connected
+# For each hidden unit within the hidden units specified above:
+for h_unit in hidden_units:
+# specify sizes of input sample (input size = X_train col size) & output sample (hidden units) in each layer
+# https://pytorch.org/docs/stable/generated/torch.nn.Linear.html#torch.nn.Linear
+ layer = nn.Linear(input_size, h_unit)
+# add each layer
+ all_layers.append(layer)
+# add activation function (trying rectified linear unit) for next layer
+ all_layers.append(nn.ReLU())
+# for the next layer to be added, the input size will be the same size as the hidden unit
+ input_size = h_unit
+
+# Specify the last layer (where input_feature = hidden_units[-1] = 3)
+all_layers.append(nn.Linear(hidden_units[-1], 1))
+
+# Set up a container that'll connect all layers in the specified sequence in the model
+model = nn.Sequential(*all_layers)
+model
This part is mainly about defining the loss function when training the model with the training data, and optimising model by using a stochastic gradient descent. One key thing I’ve gathered from trying to learn about deep learning is that we’re aiming for global minima and not local minima (e.g. if learning rate is too small, this may end up with local minima; if learning rate is too large, it may end up over-estimating the global minima). I’ve also encountered the PyTorch padding method to make sure the input and target tensors are of the same size, otherwise the model will run into matrix broadcasting issue (which will likely influence the results). The training loss appears to have converged when the epoch runs reach 100 and/or after (note this may vary due to shuffle data sampling)… (I also think my data size is way too small to show a clear contrast in training loss convergence).
+
References for: nn.MSELoss() - measures mean squared error between X and y, and nn.functional.pad() - pads tensor (increase tensor size)
+
Obtaining training loss via model training:
+
+
+Code
+
# Set up loss function
+loss_f = nn.MSELoss()
+# Set up stochastic gradient descent optimiser to optimise model (minimise loss) during training
+# lr = learning rate - default: 0.049787 (1*e^-3)
+optim = torch.optim.SGD(model.parameters(), lr=0.005)
+# Set training epochs (epoch: each cycle of training or passing through the training set)
+num_epochs =200
+# Set the log output to show training loss - for every 20 epochs
+log_epochs =20
+torch.manual_seed(1)
+# Create empty lists to save training loss (for training and testing/validation sets)
+train_epoch_loss = []
+test_epoch_loss = []
+
+# Predict via training X_batch & obtain train loss via loss function from X_batch & y_batch
+for epoch inrange(num_epochs):
+ train_loss =0
+for X_batch, y_batch in train_dl:
+# Make predictions
+ predict = model(X_batch)[:, 0]
+# Make input tensors the same size as y_batch tensors
+ predict_pad = F.pad(predict[None], pad=(1, 0, 0, 0))
+# Calculate training loss
+ loss = loss_f(predict_pad, y_batch)
+# Calculate gradients (backpropagations)
+ loss.backward(retain_graph=True)
+# Update parameters using gradients
+ optim.step()
+# Reset gradients back to zero
+ optim.zero_grad()
+ train_loss += loss.item()
+
+if epoch % log_epochs ==0:
+print(f"Epoch {epoch} Loss {train_loss/len(train_dl):.4f}")
+
+ train_epoch_loss.append(train_loss)
+
+
+
Epoch 0 Loss 4.3253
+Epoch 20 Loss 3.5549
+Epoch 40 Loss 2.9739
+Epoch 60 Loss 2.4838
+Epoch 80 Loss 2.1047
+
+
+
Epoch 100 Loss 1.8545
+Epoch 120 Loss 1.7217
+Epoch 140 Loss 1.6662
+Epoch 160 Loss 1.6471
+Epoch 180 Loss 1.6415
+
+
+
Obtaining test or validation loss:
+
+
+Code
+
# Predict via testing X_batch & obtain test loss
+for epoch inrange(num_epochs):
+ test_loss =0
+for X_batch, y_batch in test_dl:
+# Make predictions
+ predict_test = model(X_batch)[:, 0]
+# Make input tensors the same size as y_batch tensors
+ predict_pad_test = F.pad(predict_test[None], pad=(1, 0, 0, 0))
+# Calculate training loss
+ loss = loss_f(predict_pad_test, y_batch)
+# Calculate gradients (backpropagations)
+ loss.backward(retain_graph=True)
+# Update parameters using gradients
+ optim.step()
+# Reset gradients back to zero
+ optim.zero_grad()
+ test_loss += loss.item()
+
+if epoch % log_epochs ==0:
+print(f"Epoch {epoch} Loss {test_loss/len(test_dl):.4f}")
+
+ test_epoch_loss.append(test_loss)
+
+
+
Epoch 0 Loss 0.4037
+Epoch 20 Loss 0.1767
+Epoch 40 Loss 0.0963
+Epoch 60 Loss 0.0615
+Epoch 80 Loss 0.0452
+Epoch 100 Loss 0.0373
+Epoch 120 Loss 0.0335
+
+
+
Epoch 140 Loss 0.0316
+Epoch 160 Loss 0.0306
+Epoch 180 Loss 0.0301
+
+
+
+
+
+
Evaluate model
+
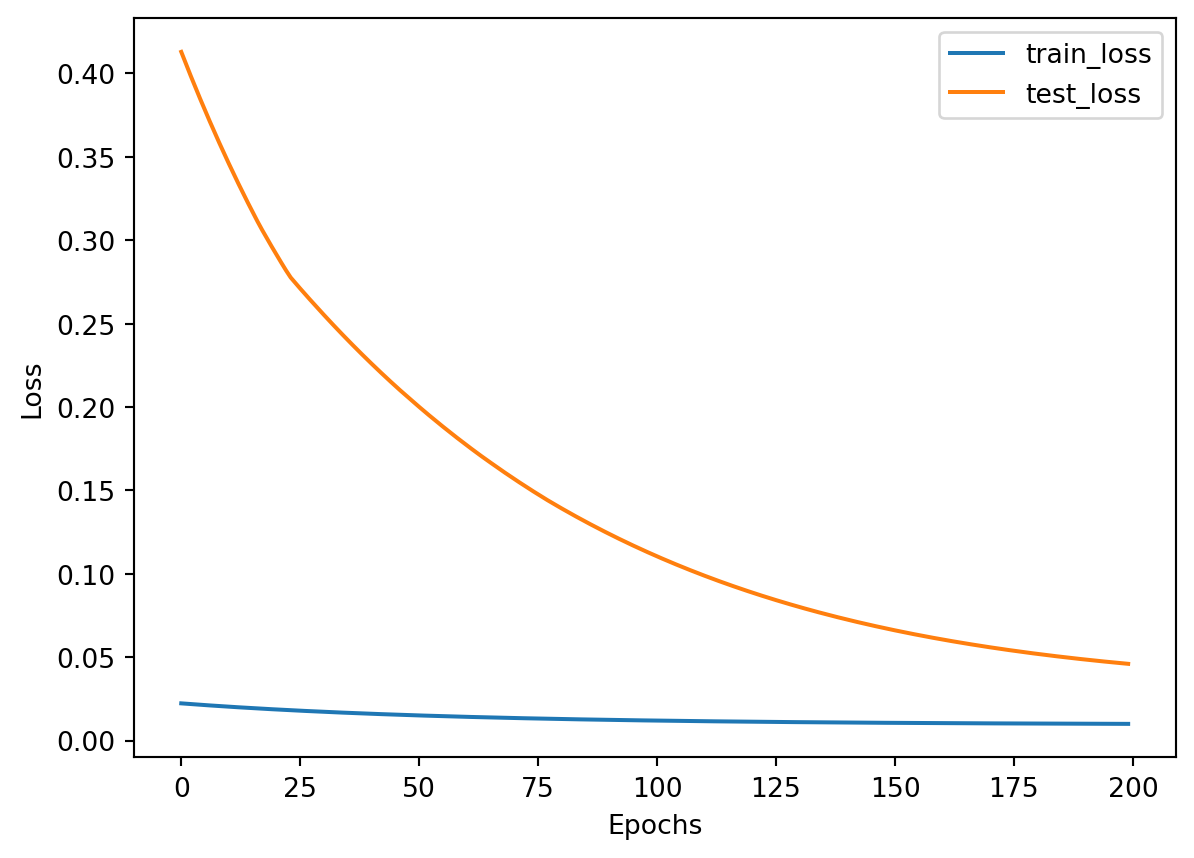
Showing train and test losses over training epochs in a plot:
At the moment, when this notebook is re-run on a refreshed kernel, this leads to a different train and test split each time, and also leading to a different train and test (validation) loss each time. There may be two types of scenarios shown in the plot above where:
+
+
test loss is higher than train loss (overfitting) - showing the model may be way too simplified and is likely under-trained
+
train loss is higher than test loss (underfitting) - showing that the model may not have been trained well, and is unable to learn the features in the training data and apply them to the test data
+
+
When there are actually more training data available with also other hyperparameters fine tuned, it may be possible to see another scenario where both test loss and train loss are very similar in trend, meaning the model is being trained well and able to generalise the training to the unseen data.
+
To mitigate overfitting:
+
+
firstly there should be more training data than what I’ve had here
+
use L1 or L2 regularisation to minimise model complexity by adding penalities to large weights
+
use early stopping during model training to stop training the model when test loss is becoming higher than the train loss
+
use torch.nn.Dropout() to randomly drop out some of the neurons to ensure the exisiting neurons will learn features without being too reliant on other neighbouring neurons in the network
+
I’ll try the early stopping or drop out method in future posts since current post is relatively long already…
+
+
To overcome underfitting:
+
+
increase training epochs
+
minimise regularisation
+
consider building a more complex or deeper neural network model
+
+
I’m trying to keep this post simple so have only used mean squared error (MSE) and mean absolute error (MAE) to evaluate the model which has made a prediction on the test set. The smaller the MSE, the less error the model has when making predictions. However this is not the only metric that will determine if a model is optimal for predictions, as I’ve also noticed that every time there’s a different train and test split, the MAE and MSE values will vary too, so it appears that some splits will generate smaller MSE and other splits will lead to larger MSE.
+
+
+Code
+
# torch.no_grad() - disable gradient calculations to reduce memory usage for inference (also like a decorator)
+with torch.no_grad():
+ predict_test = model(X_test.float())[:, 0]
+# Padding target tensor with set size of [(1, 2)] as input tensor size will vary
+# when notebook is re-run each time due to butina split with sample shuffling
+# so need to pad the target tensor accordingly
+ y_test_pad = F.pad(y_test, pad=(predict_test[None].shape[1] - y_test.shape[1], 0, 0, 0))
+ loss_new = loss_f(predict_test[None], y_test_pad)
+print(f"MSE for test set: {loss_new.item():.4f}")
+print(f"MAE for test set: {nn.L1Loss()(predict_test[None], y_test_pad).item():.4f}")
+
+
+
MSE for test set: 0.6576
+MAE for test set: 0.8070
I haven’t done feature standardisation for X_train which is to centre X_train mean and divide by its standard deviation, code may be like this, X_train_normalised = (X_train - np.mean(X_train))/np.std(X_train) (if used on training data, need to apply this to testing data too)
+
Training features are certainly too small, however, the main goal of this very first post is to get an overall idea of how to construct a baseline DNN regression model. There are lots of other things that can be done to the ADRs data e.g. adding more drug molecular features and properties. I have essentially only used the initial molecular fingerprints generated when doing the data split to add a bit of molecular aspect in the training dataset.
+
I haven’t taken into account the frequencies of words (e.g. same drug classes and same ADR terms across different drugs) in the training and testing data, however, the aim of this first piece of work is also not a semantic analysis in natural language processing so this might not be needed…
+
There may be other PyTorch functions that I do not yet know about that will deal with small datasets e.g. perhaps torch.sparse may be useful?… so this piece is certainly not the only way to do it, but one of the many ways to work with small data
+
+
+
+
+
Acknowledgements
+
I’m very thankful for the existence of these references, websites and reviewer below which have helped me understand (or scratch a small surface of) deep learning and also solve the coding issues mentioned in this post:
+
+
+
+
+ ]]>
+ Deep learning
+ Pytorch
+ RDKit
+ Pandas
+ Python
+ ChEMBL database
+ Toxicology
+ Metabolism
+ Cheminformatics
+ https://jhylin.github.io/Data_in_life_blog/posts/22_Simple_dnn_adrs/2_ADR_regressor.html
+ Tue, 07 Jan 2025 11:00:00 GMT
+Cytochrome P450 and small drug moleculesJennifer HY Lin
@@ -50663,1921 +51886,5 @@ Pedregosa, Fabian, Gaël Varoquaux, Alexandre Gramfort, Vincent Michel, Bertrand
https://jhylin.github.io/Data_in_life_blog/posts/21_ML1-1_Small_mols_in_chembl_update/ML1-1-4_chembl_cpds_evaluate.htmlTue, 03 Jan 2023 11:00:00 GMT
-
- Small molecules in ChEMBL database (old)
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/08_ML1-1_Small_molecules_in_ChEMBL_database/ML1-1_chembl_cpds.html
- This post has been updated since October 2024 (separated into four shorter posts) using only Polars dataframe library (the older version uses both Polars and Pandas):
-
*Latest update from 19th April 2024 - Polars is currently more integrated with Scikit-learn from version 1.4 (since January 2024), see this link re. Polars output in set_output for Polars dataframe outputs in Scikit-learn, and also a few other Polars enhancements from release version 1.4 changelog.
-
Previous post update was on 16th August 2023 - some code updates only, please always refer to Polars API reference documentations for most up-to-date code.
-
-
-
-
Background
-
As my interests gradually grew for Rust, I realised why so many people said it might be a hard programming language to learn. My head was spinning after reading the Rust programming language book and watching a few online teaching videos about it. I then decided to start from something I was more familiar with, and somehow through various online ventures and searching, I’ve managed to start two projects in parallel. The first one was where I used Polars dataframe library, and the second one would be about using Rust through an interactive user interface such as Jupyter notebook. I’ve anticipated that the second project would take much longer time for me to finish, so I would be tackling the first project for now.
-
This project was about using Polars, a blazingly fast dataframe library that was written completely in Rust with a very light Python binding that was available for use via Python or Rust, so I started using Polars via Python on Jupyter Lab initially, which involved data wrangling, some exploratory data analysis (EDA), and a reasonably larger section on using machine learning (ML) through scikit-learn. The editing and publishing of this post was mainly achieved via RStudio IDE.
-
-
-
-
Install Polars
-
-
# To install Polars dataframe library
-# Uncomment below to download and install Polars
-#!pip install polars
-
-# Update Polars version
-# Uncomment the line below to update Polars
-#!pip install --upgrade polars
-
-
Once Polars was installed, the next step was to import it for use.
-
-
import polars as pl
-
-
-
# Show version of Polars
-# Uncomment line below to check version of Polars installed/updated
-#pl.show_versions()
-
-
-
-
-
Download dataset
-
The dataset, which was purely about small molecules and their physicochemical properties, was downloaded from ChEMBL database and saved as a .csv file. I’ve decided not to upload the “chembl_mols.csv” file due to its sheer size (around 0.6 GB), and also I’d like to stay using free open-source resources (including GitHub) at this stage. I’ve looked into the Git large file system, but for the free version it only provides 2 GB, which at this stage, I think by adding this larger than usual .csv file along with my portfolio blog repository may exceed this limit in no time.
-
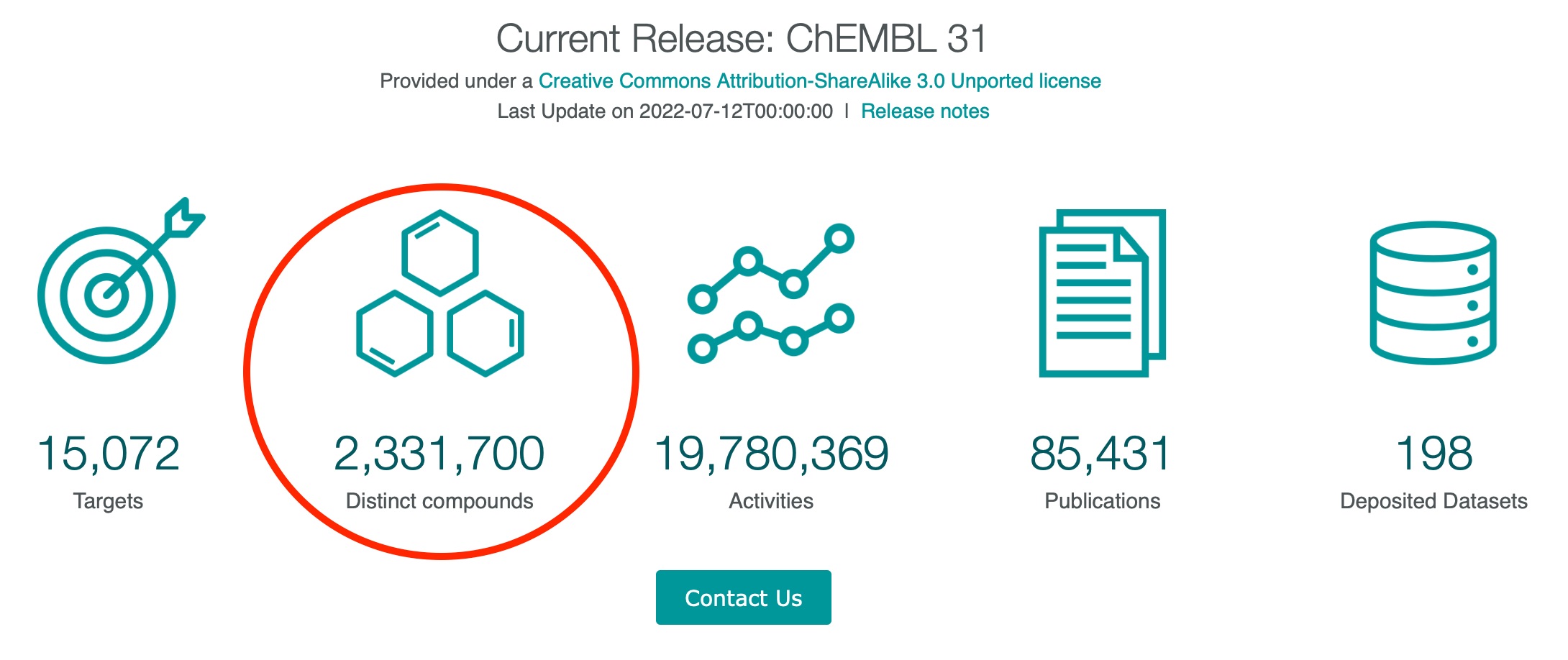
For anyone who would like to use the same dataset, the file I used would be equivalent to a straight download from the home page of ChEMBL database, via clicking on the “Distinct compounds” (please see the circled area in the image below). Options were available to download the files as .csv, .tsv or .sdf formats (located at the top right of the page).
-
-
-
-
-Image adapted from ChEMBL database website
-
-
-
-
Once we’ve had the file ready, it would be read via the usual read_csv() method.
-
-
df = pl.read_csv("chembl_mols.csv")
-df.head() #read first 5 rows
-#df #read full dataset
Now, since this dataset was downloaded as a .csv file, this meant it was likely to have a certain delimiter between each variable. So the whole dataset was presented as strings where each string represented each compound in each row. Each variable was separated by semicolons. To read it properly, I’ve added a delimiter term in the code to transform the dataframe into a more readable format.
-
-
# By referring to Polars documentation,
-# *use "sep" to set the delimiter of the file
-# which was semicolons in this case
-# *please note this has been updated to "separator"
-# due to updates in Polars since the published date of this post
-df = pl.read_csv("chembl_mols.csv", separator =";")
-# Show the first 10 rows of data
-#df.head(10)
-# or full dataset
-df
-
-
-
-shape: (2_331_700, 32)
ChEMBL ID
Name
Synonyms
Type
Max Phase
Molecular Weight
Targets
Bioactivities
AlogP
Polar Surface Area
HBA
HBD
#RO5 Violations
#Rotatable Bonds
Passes Ro3
QED Weighted
CX Acidic pKa
CX Basic pKa
CX LogP
CX LogD
Aromatic Rings
Structure Type
Inorganic Flag
Heavy Atoms
HBA (Lipinski)
HBD (Lipinski)
#RO5 Violations (Lipinski)
Molecular Weight (Monoisotopic)
Molecular Species
Molecular Formula
Smiles
Inchi Key
str
str
str
str
i64
str
str
str
str
str
str
str
str
str
str
str
str
str
str
str
str
str
i64
str
str
str
str
str
str
str
str
str
"CHEMBL1206185"
""
""
"Small molecule"
0
"607.88"
""
""
"9.46"
"89.62"
"5"
"2"
"2"
"17"
"N"
"0.09"
"-1.91"
"8.38"
"9.40"
"9.36"
"3"
"MOL"
-1
"42"
"5"
"3"
"2"
"607.2790"
"ACID"
"C35H45NO4S2"
"CCCCCCCCCCC#CC(N)c1ccccc1-c1cc…
"UFBLKYIDZFRLPR-UHFFFAOYSA-N"
"CHEMBL539070"
""
""
"Small molecule"
0
"286.79"
"1"
"1"
"2.28"
"73.06"
"6"
"2"
"0"
"5"
"N"
"0.63"
"13.84"
"3.64"
"2.57"
"2.57"
"2"
"MOL"
-1
"17"
"5"
"3"
"0"
"250.0888"
"NEUTRAL"
"C11H15ClN4OS"
"CCCOc1ccccc1-c1nnc(NN)s1.Cl"
"WPEWNRKLKLNLSO-UHFFFAOYSA-N"
"CHEMBL3335528"
""
""
"Small molecule"
0
"842.80"
"2"
"6"
"0.18"
"269.57"
"18"
"5"
"2"
"17"
"N"
"0.09"
"3.20"
"None"
"3.31"
"-0.14"
"3"
"MOL"
-1
"60"
"19"
"5"
"2"
"842.2633"
"ACID"
"C41H46O19"
"COC(=O)[C@H](O[C@@H]1O[C@@H](C…
"KGUJQZWYZPYYRZ-LWEWUKDVSA-N"
"CHEMBL2419030"
""
""
"Small molecule"
0
"359.33"
"4"
"4"
"3.94"
"85.13"
"6"
"1"
"0"
"3"
"N"
"0.66"
"None"
"None"
"3.66"
"3.66"
"2"
"MOL"
-1
"24"
"6"
"1"
"0"
"359.0551"
"NEUTRAL"
"C14H12F3N3O3S"
"O=c1nc(NC2CCCC2)sc2c([N+](=O)[…
"QGDMYSDFCXOKML-UHFFFAOYSA-N"
"CHEMBL4301448"
""
""
"Small molecule"
0
"465.55"
""
""
"5.09"
"105.28"
"6"
"4"
"1"
"10"
"N"
"0.15"
"None"
"12.14"
"4.41"
"2.00"
"4"
"MOL"
-1
"33"
"7"
"5"
"1"
"465.1635"
"BASE"
"C24H24FN5O2S"
"N=C(N)NCCCOc1ccc(CNc2nc3ccc(Oc…
"RXTJPHLPHOZLFS-UHFFFAOYSA-N"
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
"CHEMBL2017916"
""
""
"Small molecule"
0
"312.35"
"3"
"3"
"2.86"
"77.00"
"6"
"1"
"0"
"4"
"N"
"0.80"
"8.13"
"3.49"
"2.17"
"2.10"
"3"
"MOL"
-1
"22"
"6"
"1"
"0"
"312.0681"
"NEUTRAL"
"C15H12N4O2S"
"COc1ccc(-c2nnc(NC(=O)c3cccnc3)…
"XIZUJGDKNPVNQA-UHFFFAOYSA-N"
"CHEMBL374652"
""
""
"Small molecule"
0
"403.83"
"1"
"1"
"5.98"
"36.02"
"2"
"2"
"1"
"4"
"N"
"0.42"
"13.65"
"None"
"5.36"
"5.36"
"3"
"MOL"
-1
"26"
"2"
"2"
"1"
"403.0421"
"NEUTRAL"
"C18H14ClF4NOS"
"CC(O)(CSc1ccc(F)cc1)c1cc2cc(Cl…
"CRPQTBRTHURKII-UHFFFAOYSA-N"
"CHEMBL1416264"
""
""
"Small molecule"
0
"380.41"
"6"
"8"
"3.06"
"85.07"
"7"
"1"
"0"
"5"
"N"
"0.54"
"13.85"
"3.86"
"2.47"
"2.47"
"4"
"MOL"
-1
"27"
"7"
"1"
"0"
"380.0856"
"NEUTRAL"
"C18H13FN6OS"
"O=C(CSc1ccc2nnc(-c3cccnc3)n2n1…
"QVYIEKHEJKFNAT-UHFFFAOYSA-N"
"CHEMBL213734"
""
""
"Small molecule"
0
"288.26"
"2"
"3"
"2.32"
"101.70"
"5"
"2"
"0"
"5"
"N"
"0.50"
"7.20"
"None"
"2.36"
"1.95"
"2"
"MOL"
-1
"21"
"7"
"2"
"0"
"288.0746"
"NEUTRAL"
"C14H12N2O5"
"O=C(COc1ccccc1)Nc1ccc([N+](=O)…
"PZTWAHGBGTWVEB-UHFFFAOYSA-N"
"CHEMBL1531634"
""
""
"Small molecule"
0
"320.16"
"19"
"21"
"4.40"
"29.10"
"2"
"1"
"0"
"4"
"N"
"0.67"
"None"
"None"
"4.04"
"4.04"
"2"
"MOL"
-1
"19"
"2"
"1"
"0"
"319.0008"
"NEUTRAL"
"C15H11BrFNO"
"O=C(/C=C/Nc1ccc(F)cc1)c1ccc(Br…
"DKPWCCDDKFLKEC-MDZDMXLPSA-N"
-
-
-
-
Initially, I only wanted to download around 24 compounds from the ChEMBL database first. Unknowingly, I ended up downloading the whole curated set of 2,331,700 small molecules (!), and I found this out when I loaded the dataframe after setting up the delimiter for the csv file, which later led to the file size problem mentioned earlier.
-
Loading these 2,331,700 rows of data was fast, which occurred within a few seconds without exaggeration. This echoed many users’ experiences with Polars, so this was another nice surprise, and once again confirmed that Rust, and also Apache arrow, which was used as Polars’ foundation, were solid in speed.
-
Now I had the full dataframe, and I wanted to find out what types of physicochemical properties were there for the compounds.
-
-
# Print all column names and data types
-print(df.glimpse())
There were a few terms where I wasn’t sure of their exact meanings, so I went through the ChEMBL_31 schema documentation and ChEMBL database website to find out. This took a while and was an important step to take so that I would know what to do when reaching the ML phase.
-
I have selected a few physicochemical properties down below so that readers and I could gather some reasonable understandings for each term. The explanations for each term were adapted from ChEMBL_31 schema documentation (available as “Release notes” on the website), or if definitions for certain terms were not available from the documentation, I resorted to interpret them myself by going into “Dinstict compounds” section on the ChEMBL database, where I would click on, e.g. bioactivities, for a random compound in there to see what results showed up and then described them below.
-
The definitions for some of the listed physicochemical properties were:
-
Max Phase - Maximum phase of development reached for the compound (where 4 = approved). Null was where max phase has not yet been assigned.
-
Bioactivities - Various biological assays used for the compounds e.g. IC50, GI50, potency tests etc.
-
AlogP - Calculated partition coefficient
-
HBA - Number of hydrogen bond acceptors
-
HBD - Number of hydrogen bond donors
-
#RO5 Violations - Number of violations of Lipinski’s rule-of-five, using HBA and HBD definitions
-
Passes Ro3 - Indicated whether the compound passed the rule-of-three (MW < 300, logP < 3 etc)
-
QED Weighted - Weighted quantitative estimate of drug likeness (as defined by Bickerton et al., Nature Chem 2012)
-
Inorganic flag - Indicated whether the molecule was inorganic (i.e., containing only metal atoms and <2 carbon atoms), where 1 = inorganic compound and -1 = not inorganic compound (assuming 0 meant it was neither case or yet to be assigned)
-
Heavy Atoms - Number of heavy (non-hydrogen) atoms
-
CX Acidic pKa - The most acidic pKa calculated using ChemAxon v17.29.0
-
CX Basic pKa - The most basic pKa calculated using ChemAxon v17.29.0
-
CX LogP - The calculated octanol/water partition coefficient using ChemAxon v17.29.0
-
CX LogD - The calculated octanol/water distribution coefficient at pH = 7.4 using ChemAxon v17.29.0
-
Structure Type - based on compound_structures table, where SEQ indicated an entry in the protein_therapeutics table instead, NONE indicated an entry in neither tables, e.g. structure unknown
-
Inchi Key - the IUPAC international chemical identifier key
-
From the df.glimpse() method previously, there were a lot of columns with the data type of “Utf8”, which meant they were strings. There were only two columns that had “Int64”, which meant they were integers. A lot of these columns were actually storing numbers as strings. So to make my life easier, I went on to convert these data types into the more appropriate ones for selected columns.
-
-
# Convert data types for multiple selected columns
-# Note: only takes two positional arguments,
-# so needed to use [] in code to allow more than two
-
-# Multiple columns all at once - with_columns()
-# *Single column - with_column()
-# *this only worked at the time of writing the post (around published date),
-# this is not going to work currently as Polars has been updated,
-# please use with_columns() for single or multiple columns instead*
-
-# Use alias if wanting to keep original data type in column,
-# as it adds a new column under an alias name to dataframe
-df_new = df.with_columns(
- [
- (pl.col("Molecular Weight")).cast(pl.Float64, strict =False),
- (pl.col("Targets")).cast(pl.Int64, strict =False),
- (pl.col("Bioactivities")).cast(pl.Int64, strict =False),
- (pl.col("AlogP")).cast(pl.Float64, strict =False),
- (pl.col("Polar Surface Area")).cast(pl.Float64, strict =False),
- (pl.col("HBA")).cast(pl.Int64, strict =False),
- (pl.col("HBD")).cast(pl.Int64, strict =False),
- (pl.col("#RO5 Violations")).cast(pl.Int64, strict =False),
- (pl.col("#Rotatable Bonds")).cast(pl.Int64, strict =False),
- (pl.col("QED Weighted")).cast(pl.Float64, strict =False),
- (pl.col("CX Acidic pKa")).cast(pl.Float64, strict =False),
- (pl.col("CX Basic pKa")).cast(pl.Float64, strict =False),
- (pl.col("CX LogP")).cast(pl.Float64, strict =False),
- (pl.col("CX LogD")).cast(pl.Float64, strict =False),
- (pl.col("Aromatic Rings")).cast(pl.Int64, strict =False),
- (pl.col("Heavy Atoms")).cast(pl.Int64, strict =False),
- (pl.col("HBA (Lipinski)")).cast(pl.Int64, strict =False),
- (pl.col("HBD (Lipinski)")).cast(pl.Int64, strict =False),
- (pl.col("#RO5 Violations (Lipinski)")).cast(pl.Int64, strict =False),
- (pl.col("Molecular Weight (Monoisotopic)")).cast(pl.Float64, strict =False)
- ]
-)
-df_new.head()
-
-
-
-shape: (5, 32)
ChEMBL ID
Name
Synonyms
Type
Max Phase
Molecular Weight
Targets
Bioactivities
AlogP
Polar Surface Area
HBA
HBD
#RO5 Violations
#Rotatable Bonds
Passes Ro3
QED Weighted
CX Acidic pKa
CX Basic pKa
CX LogP
CX LogD
Aromatic Rings
Structure Type
Inorganic Flag
Heavy Atoms
HBA (Lipinski)
HBD (Lipinski)
#RO5 Violations (Lipinski)
Molecular Weight (Monoisotopic)
Molecular Species
Molecular Formula
Smiles
Inchi Key
str
str
str
str
i64
f64
i64
i64
f64
f64
i64
i64
i64
i64
str
f64
f64
f64
f64
f64
i64
str
i64
i64
i64
i64
i64
f64
str
str
str
str
"CHEMBL1206185"
""
""
"Small molecule"
0
607.88
null
null
9.46
89.62
5
2
2
17
"N"
0.09
-1.91
8.38
9.4
9.36
3
"MOL"
-1
42
5
3
2
607.279
"ACID"
"C35H45NO4S2"
"CCCCCCCCCCC#CC(N)c1ccccc1-c1cc…
"UFBLKYIDZFRLPR-UHFFFAOYSA-N"
"CHEMBL539070"
""
""
"Small molecule"
0
286.79
1
1
2.28
73.06
6
2
0
5
"N"
0.63
13.84
3.64
2.57
2.57
2
"MOL"
-1
17
5
3
0
250.0888
"NEUTRAL"
"C11H15ClN4OS"
"CCCOc1ccccc1-c1nnc(NN)s1.Cl"
"WPEWNRKLKLNLSO-UHFFFAOYSA-N"
"CHEMBL3335528"
""
""
"Small molecule"
0
842.8
2
6
0.18
269.57
18
5
2
17
"N"
0.09
3.2
null
3.31
-0.14
3
"MOL"
-1
60
19
5
2
842.2633
"ACID"
"C41H46O19"
"COC(=O)[C@H](O[C@@H]1O[C@@H](C…
"KGUJQZWYZPYYRZ-LWEWUKDVSA-N"
"CHEMBL2419030"
""
""
"Small molecule"
0
359.33
4
4
3.94
85.13
6
1
0
3
"N"
0.66
null
null
3.66
3.66
2
"MOL"
-1
24
6
1
0
359.0551
"NEUTRAL"
"C14H12F3N3O3S"
"O=c1nc(NC2CCCC2)sc2c([N+](=O)[…
"QGDMYSDFCXOKML-UHFFFAOYSA-N"
"CHEMBL4301448"
""
""
"Small molecule"
0
465.55
null
null
5.09
105.28
6
4
1
10
"N"
0.15
null
12.14
4.41
2.0
4
"MOL"
-1
33
7
5
1
465.1635
"BASE"
"C24H24FN5O2S"
"N=C(N)NCCCOc1ccc(CNc2nc3ccc(Oc…
"RXTJPHLPHOZLFS-UHFFFAOYSA-N"
-
-
-
Once all the columns’ data types have been checked and converted to appropriate types accordingly, I used null_count() to see the distributions of all null entries in the dataset.
-
-
# Check for any null or NA or "" entries in the dataset
-# Alternative code that worked similarly was df.select(pl.all().null_count())
-df_new.null_count()
-
-
-
-shape: (1, 32)
ChEMBL ID
Name
Synonyms
Type
Max Phase
Molecular Weight
Targets
Bioactivities
AlogP
Polar Surface Area
HBA
HBD
#RO5 Violations
#Rotatable Bonds
Passes Ro3
QED Weighted
CX Acidic pKa
CX Basic pKa
CX LogP
CX LogD
Aromatic Rings
Structure Type
Inorganic Flag
Heavy Atoms
HBA (Lipinski)
HBD (Lipinski)
#RO5 Violations (Lipinski)
Molecular Weight (Monoisotopic)
Molecular Species
Molecular Formula
Smiles
Inchi Key
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
0
0
0
0
0
23249
96223
96223
83571
83571
83571
83571
83571
83571
0
83571
1052439
882168
83795
83795
83571
0
0
83571
83571
83571
83571
23252
0
0
0
0
-
-
-
-
# Drop rows with null entries
-df_dn = df_new.drop_nulls()
-df_dn
-# Number of rows reduced to 736,570
-
-
-
-shape: (736_570, 32)
ChEMBL ID
Name
Synonyms
Type
Max Phase
Molecular Weight
Targets
Bioactivities
AlogP
Polar Surface Area
HBA
HBD
#RO5 Violations
#Rotatable Bonds
Passes Ro3
QED Weighted
CX Acidic pKa
CX Basic pKa
CX LogP
CX LogD
Aromatic Rings
Structure Type
Inorganic Flag
Heavy Atoms
HBA (Lipinski)
HBD (Lipinski)
#RO5 Violations (Lipinski)
Molecular Weight (Monoisotopic)
Molecular Species
Molecular Formula
Smiles
Inchi Key
str
str
str
str
i64
f64
i64
i64
f64
f64
i64
i64
i64
i64
str
f64
f64
f64
f64
f64
i64
str
i64
i64
i64
i64
i64
f64
str
str
str
str
"CHEMBL539070"
""
""
"Small molecule"
0
286.79
1
1
2.28
73.06
6
2
0
5
"N"
0.63
13.84
3.64
2.57
2.57
2
"MOL"
-1
17
5
3
0
250.0888
"NEUTRAL"
"C11H15ClN4OS"
"CCCOc1ccccc1-c1nnc(NN)s1.Cl"
"WPEWNRKLKLNLSO-UHFFFAOYSA-N"
"CHEMBL3827271"
""
""
"Small molecule"
0
712.85
1
1
-2.84
319.06
10
11
2
16
"N"
0.07
4.08
10.49
-6.88
-8.95
0
"MOL"
-1
50
19
14
3
712.4232
"ZWITTERION"
"C31H56N10O9"
"CC(C)C[C@@H]1NC(=O)[C@H](CCCNC…
"QJQNNLICZLLPMB-VUBDRERZSA-N"
"CHEMBL3824158"
""
""
"Small molecule"
0
422.48
2
4
5.09
109.54
6
2
1
10
"N"
0.31
4.59
7.99
2.49
2.42
2
"MOL"
-1
31
7
2
1
422.1842
"ACID"
"C24H26N2O5"
"CCCCCCCNC(C1=C(O)C(=O)c2ccccc2…
"AXOVDUYYBUYLPC-UHFFFAOYSA-N"
"CHEMBL1991010"
""
""
"Small molecule"
0
454.05
60
60
5.18
40.54
3
1
1
8
"N"
0.6
13.88
8.48
6.34
5.22
2
"MOL"
-1
31
3
1
1
417.2668
"NEUTRAL"
"C28H36ClNO2"
"CCc1ccc(/C=C/C(=O)C2CN(CC)CCC2…
"XJDPAUYFONOZBC-DCPGAFKKSA-N"
"CHEMBL195644"
""
""
"Small molecule"
0
375.47
2
3
4.95
70.42
4
2
0
2
"N"
0.73
9.52
3.73
3.92
3.91
2
"MOL"
-1
28
4
2
0
375.1834
"NEUTRAL"
"C24H25NO3"
"C[C@]12CCC3c4ccc(O)cc4CCC3C1CC…
"MOBPUUUBXAHZBM-KSAYNYSMSA-N"
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
…
"CHEMBL2419480"
""
""
"Small molecule"
0
456.52
3
3
1.56
129.46
8
1
0
8
"N"
0.59
3.99
1.9
2.14
1.2
2
"MOL"
-1
32
9
1
0
456.1467
"ACID"
"C22H24N4O5S"
"CCOC(=O)c1cc(C#N)c(N2CC(C(=O)N…
"TXYSLOQUANFYQS-UHFFFAOYSA-N"
"CHEMBL540121"
""
""
"Small molecule"
0
540.05
2
3
2.39
147.14
6
4
1
8
"N"
0.22
5.02
11.48
-0.75
-0.78
4
"MOL"
-1
36
9
5
1
503.1627
"ZWITTERION"
"C26H26ClN5O4S"
"Cc1ccn(NS(=O)(=O)c2cccc3ccccc2…
"TZLGWENJAJXWGA-UHFFFAOYSA-N"
"CHEMBL374041"
""
""
"Small molecule"
0
504.5
2
4
3.04
144.95
8
3
1
10
"N"
0.28
6.59
4.37
2.17
1.33
3
"MOL"
-1
37
11
3
2
504.1645
"NEUTRAL"
"C26H24N4O7"
"CCOCCC1(Oc2ccc(Oc3ccc(C(=O)Nc4…
"ABCSNHDQYHOLOO-UHFFFAOYSA-N"
"CHEMBL2017916"
""
""
"Small molecule"
0
312.35
3
3
2.86
77.0
6
1
0
4
"N"
0.8
8.13
3.49
2.17
2.1
3
"MOL"
-1
22
6
1
0
312.0681
"NEUTRAL"
"C15H12N4O2S"
"COc1ccc(-c2nnc(NC(=O)c3cccnc3)…
"XIZUJGDKNPVNQA-UHFFFAOYSA-N"
"CHEMBL1416264"
""
""
"Small molecule"
0
380.41
6
8
3.06
85.07
7
1
0
5
"N"
0.54
13.85
3.86
2.47
2.47
4
"MOL"
-1
27
7
1
0
380.0856
"NEUTRAL"
"C18H13FN6OS"
"O=C(CSc1ccc2nnc(-c3cccnc3)n2n1…
"QVYIEKHEJKFNAT-UHFFFAOYSA-N"
-
-
-
-
# Check that all rows with null values were dropped
-df_dn.null_count()
-
-
-
-shape: (1, 32)
ChEMBL ID
Name
Synonyms
Type
Max Phase
Molecular Weight
Targets
Bioactivities
AlogP
Polar Surface Area
HBA
HBD
#RO5 Violations
#Rotatable Bonds
Passes Ro3
QED Weighted
CX Acidic pKa
CX Basic pKa
CX LogP
CX LogD
Aromatic Rings
Structure Type
Inorganic Flag
Heavy Atoms
HBA (Lipinski)
HBD (Lipinski)
#RO5 Violations (Lipinski)
Molecular Weight (Monoisotopic)
Molecular Species
Molecular Formula
Smiles
Inchi Key
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
u32
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
0
-
-
-
-
# To see summary statistics for df_dn dataset
-df_dn.describe()
-
-
-
-shape: (9, 33)
statistic
ChEMBL ID
Name
Synonyms
Type
Max Phase
Molecular Weight
Targets
Bioactivities
AlogP
Polar Surface Area
HBA
HBD
#RO5 Violations
#Rotatable Bonds
Passes Ro3
QED Weighted
CX Acidic pKa
CX Basic pKa
CX LogP
CX LogD
Aromatic Rings
Structure Type
Inorganic Flag
Heavy Atoms
HBA (Lipinski)
HBD (Lipinski)
#RO5 Violations (Lipinski)
Molecular Weight (Monoisotopic)
Molecular Species
Molecular Formula
Smiles
Inchi Key
str
str
str
str
str
f64
f64
f64
f64
f64
f64
f64
f64
f64
f64
str
f64
f64
f64
f64
f64
f64
str
f64
f64
f64
f64
f64
f64
str
str
str
str
"count"
"736570"
"736570"
"736570"
"736570"
736570.0
736570.0
736570.0
736570.0
736570.0
736570.0
736570.0
736570.0
736570.0
736570.0
"736570"
736570.0
736570.0
736570.0
736570.0
736570.0
736570.0
"736570"
736570.0
736570.0
736570.0
736570.0
736570.0
736570.0
"736570"
"736570"
"736570"
"736570"
"null_count"
"0"
"0"
"0"
"0"
0.0
0.0
0.0
0.0
0.0
0.0
0.0
0.0
0.0
0.0
"0"
0.0
0.0
0.0
0.0
0.0
0.0
"0"
0.0
0.0
0.0
0.0
0.0
0.0
"0"
"0"
"0"
"0"
"mean"
null
null
null
null
0.007937
431.880042
5.520715
8.705471
3.325204
97.58116
5.890221
2.274721
0.489124
6.216262
null
0.510936
9.59944
5.074377
2.815115
2.17363
2.754412
null
-0.929521
30.266113
7.276555
2.497847
0.576319
428.334452
null
null
null
null
"std"
null
null
null
null
0.164565
135.637543
14.784793
55.537836
1.980414
47.40847
2.459106
1.681943
0.794171
3.894505
null
0.229039
3.583639
3.234099
2.286325
2.645694
1.2009
null
0.255953
9.54406
3.067158
2.081485
0.908719
133.755653
null
null
null
null
"min"
"CHEMBL10"
""
""
""
0.0
45.04
1.0
1.0
-12.92
3.24
1.0
0.0
0.0
0.0
"N"
0.01
-20.03
0.0
-16.71
-26.04
0.0
"BOTH"
-1.0
3.0
1.0
0.0
0.0
45.0215
"ACID"
"C10H10Br2N2O"
"Br.Br.C/C(=N/NC(=N)N)c1ccc(CNC…
"AAAADVYFXUUVEO-UHFFFAOYSA-N"
"25%"
null
null
null
null
0.0
340.47
1.0
2.0
2.21
68.29
4.0
1.0
0.0
4.0
null
0.34
7.69
2.25
1.64
0.97
2.0
null
-1.0
24.0
5.0
1.0
0.0
339.0582
null
null
null
null
"50%"
null
null
null
null
0.0
413.46
2.0
3.0
3.37
88.32
5.0
2.0
0.0
5.0
null
0.51
10.51
4.7
2.97
2.46
3.0
null
-1.0
29.0
7.0
2.0
0.0
410.2066
null
null
null
null
"75%"
null
null
null
null
0.0
494.57
5.0
8.0
4.53
113.23
7.0
3.0
1.0
8.0
null
0.7
12.45
7.85
4.23
3.79
4.0
null
-1.0
35.0
9.0
3.0
1.0
491.0028
null
null
null
null
"max"
"CHEMBL99998"
"t-4-AMINOCROTONIC ACID (TACA)"
"trovafloxacin9"
"Unknown"
4.0
1901.51
1334.0
17911.0
16.83
595.22
32.0
25.0
4.0
59.0
"Y"
0.95
14.0
38.8
18.31
18.31
28.0
"MOL"
0.0
76.0
34.0
32.0
4.0
999.4063
"ZWITTERION"
"HNNa2O8S2"
"n1nc2c([nH]1)c1nn[nH]c1c1nn[nH…
"ZZZZEJJXQQRZBH-UHFFFAOYSA-N"
-
-
-
-
-
-
Some exploratory data analysis
-
One of the columns that jumped out from the summary statistics of the df_dn dataset was the “Targets” column. It ranged from 1 to 1334 targets. Out of curiosity, I went through several places on ChEMBL website to find out the exact definition of “Target”. Eventually I settled on an answer which explained that the “Target” column represented the number of targets associated with the particular ChEMBL compound listed. I then singled out the ChEMBL compound with 1334 targets recorded, it turned out to be imatinib, which was marketed as Gleevec, and was a well-known prescription medicine for leukaemia and other selected oncological disorders with many well-documented drug interactions.
-
-
# This was confirmed via a filter function, which brought up CHEMBL1421, or also known as dasatinib
-df_dn.filter(pl.col("Targets") ==1334)
-
-
-
-shape: (1, 32)
ChEMBL ID
Name
Synonyms
Type
Max Phase
Molecular Weight
Targets
Bioactivities
AlogP
Polar Surface Area
HBA
HBD
#RO5 Violations
#Rotatable Bonds
Passes Ro3
QED Weighted
CX Acidic pKa
CX Basic pKa
CX LogP
CX LogD
Aromatic Rings
Structure Type
Inorganic Flag
Heavy Atoms
HBA (Lipinski)
HBD (Lipinski)
#RO5 Violations (Lipinski)
Molecular Weight (Monoisotopic)
Molecular Species
Molecular Formula
Smiles
Inchi Key
str
str
str
str
i64
f64
i64
i64
f64
f64
i64
i64
i64
i64
str
f64
f64
f64
f64
f64
i64
str
i64
i64
i64
i64
i64
f64
str
str
str
str
"CHEMBL941"
"IMATINIB"
"GLAMOX|Gleevec|IMATINIB|Imatin…
"Small molecule"
4
493.62
1334
4359
4.59
86.28
7
2
0
7
"N"
0.39
12.69
7.84
4.38
3.8
4
"MOL"
0
37
8
2
0
493.259
"NEUTRAL"
"C29H31N7O"
"Cc1ccc(NC(=O)c2ccc(CN3CCN(C)CC…
"KTUFNOKKBVMGRW-UHFFFAOYSA-N"
-
-
-
To explore other physicochemical and molecular properties in the dataframe, “Max Phase” was one of the first few that drew my interests. So it tagged each ChEMBL compound with a max phase number from 0 to 4, where 4 meant the compound was approved (usually also meant it was already a prescription medicine). Thinking along this line, I thought what about those compounds that had max phase as 0, because they were the ones still pending associations with max phase numbers. By extending on this idea, this could be a good opportunity to introduce some ML to predict whether these zero max phase compounds would enter the approved max phase.
-
Firstly, I had a look at the overall distribution of the max phase compounds in this dataframe df_dn.
-
-
# Interested in what types of "Max Phase" were recorded
-# for the curated small molecules in ChEMBL database
-df_dn.group_by("Max Phase", maintain_order =True).agg(pl.count())
-
-
/var/folders/0y/p72zn_cx4vz1lv6zmyd7gkt00000gn/T/ipykernel_1534/3278305966.py:3: DeprecationWarning: `pl.count()` is deprecated. Please use `pl.len()` instead.
- df_dn.group_by("Max Phase", maintain_order = True).agg(pl.count())
-
-
-
-
-shape: (5, 2)
Max Phase
count
i64
u32
0
734633
3
303
4
954
2
441
1
239
-
-
-
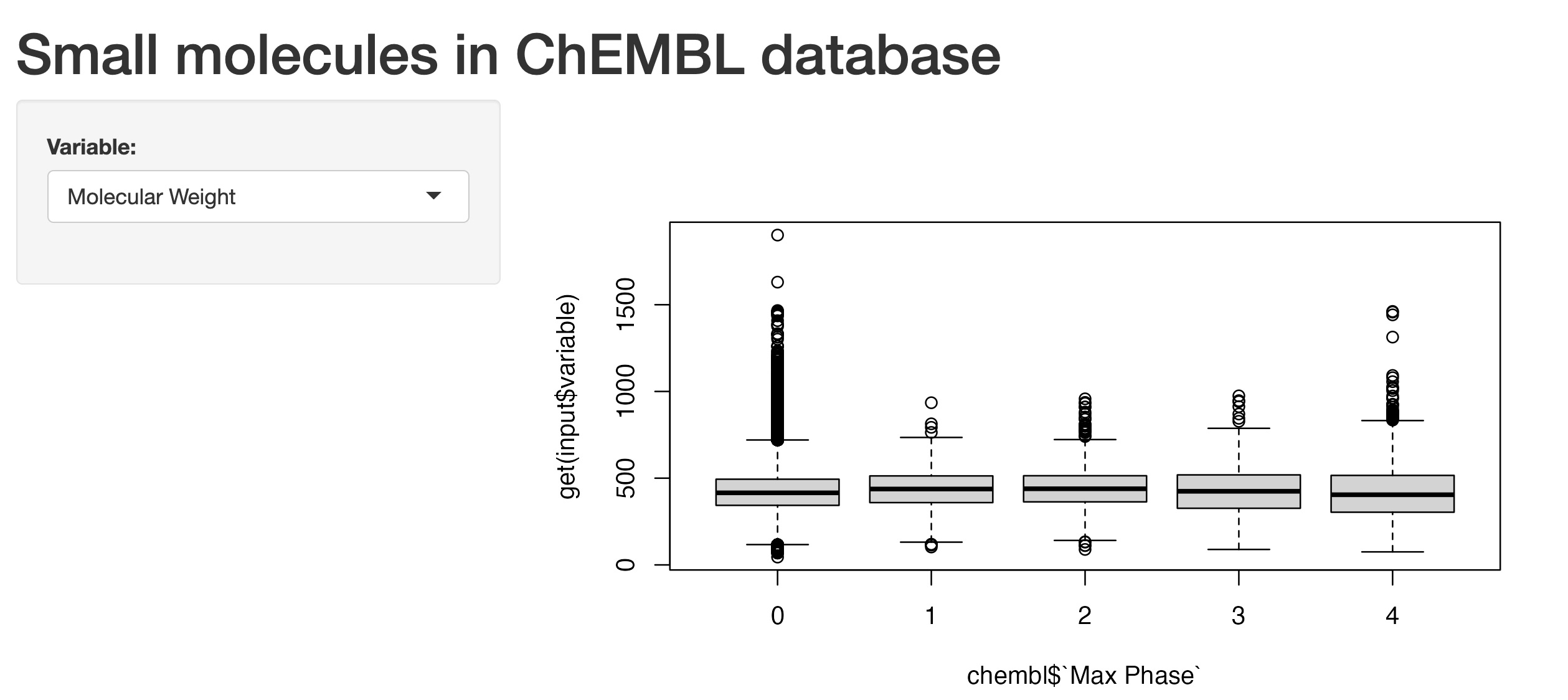
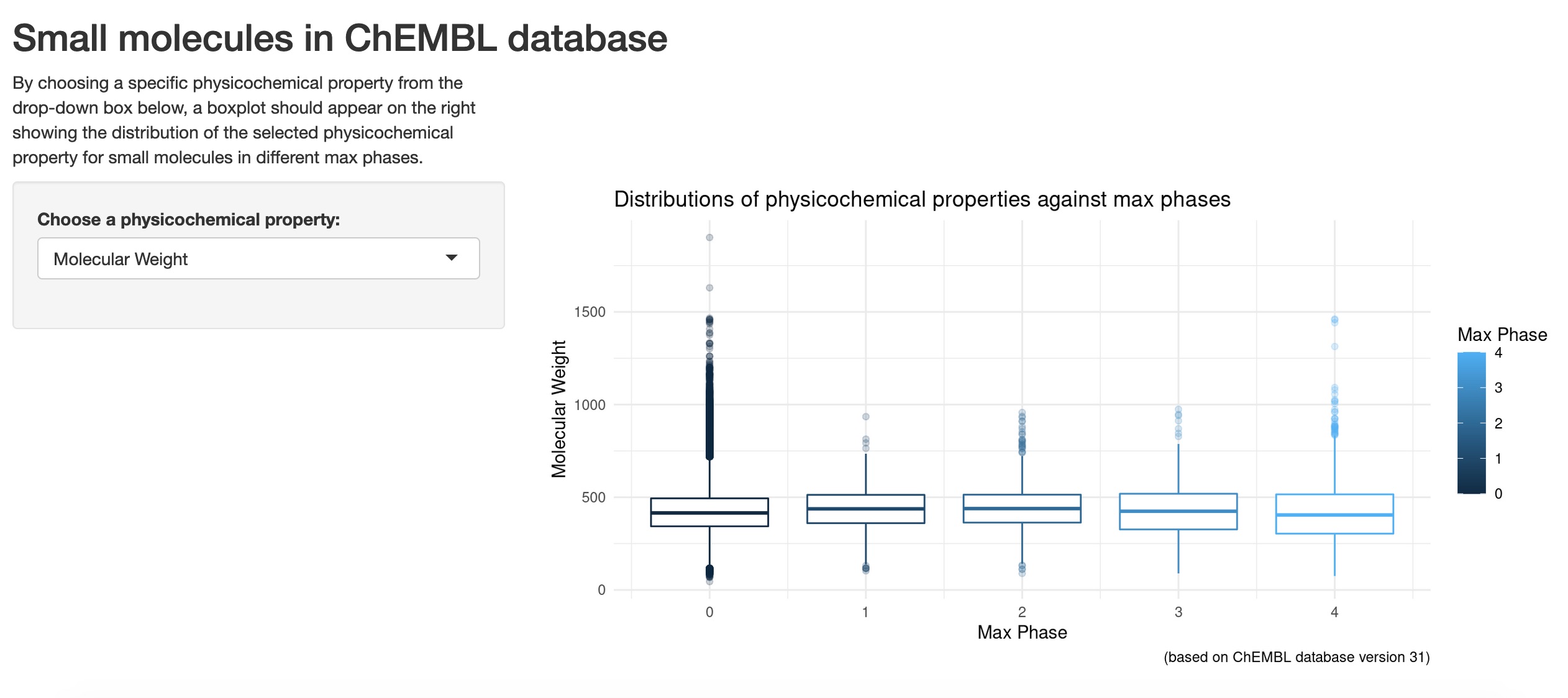
A quick groupby function showed that there were only 954 small molecules approved. Phase 3 recorded a total of 303 small molecules. For phase 2, there were 441 small molecules, followed by 239 compounds in phase 1. There were, however, a total amount of 734,633 small molecules that had zero as phase number (as per ChEMBL_31 schema documentation). Note: these figures were only for ChEMBL compounds with full documentations in the dataset (excluding entries or compounds with N/A or “” (empty) string cells).
-
One of the other parameters I was interested in was “QED Weighted”. So I went further into understanding what it meant, as the original reference was conveniently provided in the ChEMBL_31 schema documentation. The reference paper was by Bickerton, G., Paolini, G., Besnard, J. et al. Quantifying the chemical beauty of drugs. Nature Chem 4, 90–98 (2012) (note: author’s manuscript was available to view via PubMed link, the Nature Chemistry link only provided abstract with access to article via other means as stated).
-
In short, it was a measure of druglikeness for small molecules based on the concept of desirability, which was based on a total of 8 different molecular properties. These molecular properties included molecular weight, ALogP, polar surface area, number of hydrogen bond acceptors, number of hydrogen bond donors, number of rotatable bonds, number of aromatic rings and structural alerts. Without going into too much details for this QED Weighted parameter, it was normally recorded as a number that ranged from 0 to 1, with 0 being the least druglike and 1 being the most druglike.
-
-
-
-
Prepare dataframe prior to running machine learning model
-
Before I got too carried away with further EDA, I wanted to get started on preparing a dataframe for the ML model. A rough plan at this stage was to filter out Max Phase 4 and 0 compounds. Max phase 0 compounds were the ones that were not assigned with any max phase numbers yet, so they would be ideal for use as the testing set. Another main idea was to use “Max Phase” parameter as the target y variable for a LR model, because ultimately stakeholders would be more interested in knowing which candidate compounds had the most likely chance to reach the final approved phase during a drug discovery and development project or otherwise. This would also provide a chance to potentially reduce the amount of resources and time required in such a complex and sophisticated matter.
-
The goal of this ML model was to answer this question: which physicochemical parameters would be the most suitable ones to predict whether a compound would enter max phase 4 (approved) or not? (implicitly, this might also help to predict which max phase 0 compounds would likely enter max phase 4 in the end)
-
I’ve then narrowed down the df_dn dataset to fulfill the following criteria:
-
-
Only small molecules present
-
Max phase of 0 and 4 only
-
-
Another reason behind choosing only small molecules that had max phase of 0 and 4 was that a confusion matrix could be built in the end to see if the parameters selected would give us a reasonably good model for predicting the outcomes of these small molecules.
-
For now, I’ve chosen the following columns (or physicochemical parameters) to appear in the interim df_0 and df_4 datasets.
Because of the large number of Max Phase 0 compounds present in the original dataset, I’ve randomly sampled about 950 small molecules from this group, so that there were similar amount of data in each group to avoid having an imbalanced dataset.
Since the plan was to use LR method for ML model, the y variable I was interested in was going to be a binary categorical variable - meaning it needed to be 0 (not approved) or 1 (approved). To do this, I’ve added a new column with a new name of “Max_Phase” and replace “4” as “1” by dividing the whole column by 4 to reach this new label.
Then I changed the data type of “Max_Phase” from float to integer, so that the two different dataframes could be concatenated (which would only work if both were of same data types).
Also I’ve created a new column with the same name of “Max_Phase” for Max phase 0 small molecules, so that the two dataframes could be combined (also needed to have exactly the same column names for it to work).
This df_concat dataset was checked to see it had all compounds in Max Phase 0 and 4 only. Note: Max Phase 4 (approved) compounds were re-labelled as Max_Phase = 1.
-
-
df_concat.group_by("Max_Phase").count()
-
-
/var/folders/0y/p72zn_cx4vz1lv6zmyd7gkt00000gn/T/ipykernel_1534/631682218.py:1: DeprecationWarning: `GroupBy.count` is deprecated. It has been renamed to `len`.
- df_concat.group_by("Max_Phase").count()
-
-
-
-
-shape: (2, 2)
Max_Phase
count
i64
u32
0
950
1
944
-
-
-
I then checked df_concat dataset only had small molecules to confirm what I’ve tried to achieve.
-
-
df_concat.group_by("Type").count()
-
-
/var/folders/0y/p72zn_cx4vz1lv6zmyd7gkt00000gn/T/ipykernel_1534/3888448416.py:1: DeprecationWarning: `GroupBy.count` is deprecated. It has been renamed to `len`.
- df_concat.group_by("Type").count()
-
-
-
-
-shape: (1, 2)
Type
count
str
u32
"Small molecule"
1894
-
-
-
So here we had the final version of the dataset, which I’ve renamed to df_ml to avoid confusion from the previous dataframes, before entering the ML phase.
-
-
# Leave out ChEMBL ID and Type
-df_ml = df_concat.select(["Max_Phase",
-"#RO5 Violations",
-"QED Weighted",
-"CX LogP",
-"CX LogD",
-"Heavy Atoms"]
- )
-df_ml
-
-
-
-shape: (1_894, 6)
Max_Phase
#RO5 Violations
QED Weighted
CX LogP
CX LogD
Heavy Atoms
i64
i64
f64
f64
f64
i64
0
0
0.66
1.25
1.24
20
0
0
0.71
3.29
3.28
17
0
1
0.38
1.29
1.28
37
0
2
0.29
3.27
3.13
45
0
0
0.55
3.52
3.52
34
…
…
…
…
…
…
1
2
0.1
5.57
5.57
65
1
0
0.83
3.3
2.61
27
1
0
0.26
3.6
1.93
32
1
0
0.77
2.43
2.43
24
1
0
0.49
2.09
1.86
34
-
-
-
-
# Check for any nulls in the dataset
-df_ml.null_count()
-
-
-
-shape: (1, 6)
Max_Phase
#RO5 Violations
QED Weighted
CX LogP
CX LogD
Heavy Atoms
u32
u32
u32
u32
u32
u32
0
0
0
0
0
0
-
-
-
-
# Check data types in df_ml dataset
-# Needed to be integers or floats for scikit-learn algorithms to work
-df_ml.dtypes
-
-
[Int64, Int64, Float64, Float64, Float64, Int64]
-
-
-
```{python}
-# Note: exported df_ml dataframe as csv file for ML series 1.2.
-df_ml.write_csv("df_ml.csv", sep =",")
-```
-
-
-
-
-
Import libraries for machine learning
-
-
# Install scikit-learn - an open-source ML library
-# Uncomment the line below if needing to install this library
-#!pip install -U scikit-learn
-
-
-
# Import scikit-learn
-import sklearn
-
-# Check version of scikit-learn
-print(sklearn.__version__)
-
-
1.5.0
-
-
-
Other libraries needed to generate ML model were imported as below.
-
-
# To use NumPy arrays to prepare X & y variables
-import numpy as np
-
-# Needed for dataframe in scikit-learn ML
-# Uncomment line below if requiring to install pandas
-#!pip install pandas
-import pandas as pd
-
-# To normalise dataset prior to running ML
-from sklearn import preprocessing
-# To split dataset into training & testing sets
-from sklearn.model_selection import train_test_split
-
-# For data visualisations
-# Uncomment line below if requiring to install matplotlib
-#!pip install matplotlib
-import matplotlib.pyplot as plt
-
-
I’ve then installed pyarrow, to convert Polars dataframe into a Pandas dataframe, which was needed to run scikit-learn.
-
-
# Uncomment line below to install pyarrow
-#!pip install pyarrow
LR was one of the supervised methods in statistical ML realm. As the term “supervised” suggested, this type of ML was purely data-driven to allow computers to learn patterns from input data with known outcomes, in order to predict new outcomes on novel data.
# Define y variable
-# Note to use "Max_Phase", not the original "Max Phase"
-y = np.asarray(df_ml_pd["Max_Phase"])
-y[0:5]
-
-
array([0, 0, 0, 0, 0])
-
-
-
-
-
-
Training and testing sets
-
-
# Split dataset into training & testing sets
-
-# Random number generator
-#rng = np.random.RandomState(0) - note: this may produce different result each time
-
-# Edited post to use random_state = 250 to show comparison with ML series 1.2
-# for reproducible result
-X_train, X_test, y_train, y_test = train_test_split(X, y, test_size =0.2, random_state =250)
-print('Training set:', X_train.shape, y_train.shape)
-print('Testing set:', X_test.shape, y_test.shape)
-
-
Training set: (1515, 5) (1515,)
-Testing set: (379, 5) (379,)
-
-
-
-
-
-
Preprocessing data
-
-
# Normalise & clean the dataset
-# Fit on the training set - not on testing set as this might lead to data leakage
-# Transform on the testing set
-X = preprocessing.StandardScaler().fit(X_train).transform(X_test)
-X[0:5]
# Import logistic regression
-from sklearn.linear_model import LogisticRegression
-# Create an instance of logistic regression classifier and fit the data
-LogR = LogisticRegression().fit(X_train, y_train)
-LogR
-
-
LogisticRegression()
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
LogisticRegression()
-
-
-
-
-
-
Applying LR classifier on testing set for prediction
# Predicted values were based on log odds
-# Use describe() method to get characteristics of the distribution
-pred = pd.DataFrame(LogR.predict_log_proba(X))
-pred.describe()
-
-
-
-
-
-
-
-
-
0
-
1
-
-
-
-
-
count
-
379.000000
-
379.000000
-
-
-
mean
-
-0.862452
-
-0.616678
-
-
-
std
-
0.318816
-
0.258353
-
-
-
min
-
-1.622114
-
-1.537892
-
-
-
25%
-
-1.096951
-
-0.775959
-
-
-
50%
-
-0.826656
-
-0.575382
-
-
-
75%
-
-0.616677
-
-0.406300
-
-
-
max
-
-0.241859
-
-0.219999
-
-
-
-
-
-
-
Alternatively, a quicker way to get predicted probabilities was via predict_proba() method in scikit-learn.
-
-
y_mp_proba = LogR.predict_proba(X_test)
-# Uncomment below to see the predicted probabilities printed
-#print(y_mp_proba)
-
-
-
-
-
Converting predicted probabilities into a dataframe
-
-
# Use describe() to show distributions
-y_mp_prob = pd.DataFrame(y_mp_proba)
-y_mp_prob.describe()
-
-
-
-
-
-
-
-
-
0
-
1
-
-
-
-
-
count
-
379.000000
-
379.000000
-
-
-
mean
-
0.482996
-
0.517004
-
-
-
std
-
0.169226
-
0.169226
-
-
-
min
-
0.008877
-
0.151562
-
-
-
25%
-
0.365433
-
0.393816
-
-
-
50%
-
0.504758
-
0.495242
-
-
-
75%
-
0.606184
-
0.634567
-
-
-
max
-
0.848438
-
0.991123
-
-
-
-
-
-
-
-
-
-
Pipeline method for LR
-
This was something I thought to try when I was reading through scikit-learn documentation. One major advantage of using pipeline was that it was designed to chain all the estimators used for ML. The benefit of this was that we only had to call fit and predict once in our data to fit the whole chain of estimators. The other useful thing was that this could avoid data leakage from our testing set into the training set by making sure the same set of samples were used to train the transformers and predictors. One other key thing it also helped was that it also avoided the possibility of missing out on the transformation step.
-
The example below used the function of make_pipeline, which took in a number of estimators as inputted, and then constructed a pipeline based on them.
-
-
# Test pipline from scikit-Learn
-from sklearn.pipeline import make_pipeline
-from sklearn.preprocessing import StandardScaler
-
-LR = make_pipeline(StandardScaler(), LogisticRegression())
-LR.fit(X_train, y_train)
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
from sklearn.metrics import accuracy_score
-accuracy_score(y_mp, y_test)
-
-
0.7018469656992085
-
-
-
The accuracy score was 0.7 (after rounding up) based on the original data preprocessing method, which meant that there were around 70% of the cases (or compounds) classified correctly by using this LR classifier. Accuracy literally provided a measure of how close the predicted samples were to the true values. One caveat to note was that for imbalanced dataset, accuracy score might not be very informative, and other evaluation metrics would need to be considered instead.
-
The accuracy score shown below was from the pipeline method used previously, which showed a very similar accuracy score of 0.69656992 (close to 0.7), confirming the method was in line with the original preprocessing method.
-
-
LR.score(X_test, y_test)
-
-
0.6965699208443272
-
-
-
-
-
-
Confusion matrix
-
Next, I’ve built a confusion matrix based on the model in order to visualise the counts of correct and incorrect predictions. The function code used below was adapted from the IBM data science course I’ve taken around the end of last year. I’ve added comments to try and explain what each section of the code meant.
-
-
# Import confusion matrix from scikit-learn
-from sklearn.metrics import confusion_matrix
-# Import itertools - functions to create iterators for efficient looping
-import itertools
-
-# Function to print and plot confusion matrix
-def plot_confusion_matrix(# Sets a cm object (cm = confusion matrix)
- cm,
-# Sets classes of '1s' (Successes) & '0s' (Non-successes) for the cm
- classes,
-# If setting normalize = true, reports in ratios instead of counts
- normalize,
- title ='Confusion matrix',
-# Choose colour of the cm (using colourmap recognised by matplotlib)
- cmap = plt.cm.Reds):
-
-if normalize:
- cm = cm.astype('float') / cm.sum(axis =1)[:, np.newaxis]
-print("Normalized confusion matrix")
-else:
-print('Confusion matrix, without normalization')
-
-print(cm)
-
-# Plot the confusion matrix
- plt.imshow(cm, interpolation ='nearest', cmap = cmap)
- plt.title(title)
- plt.colorbar()
- tick_marks = np.arange(len(classes))
- plt.xticks(tick_marks, classes, rotation =45)
- plt.yticks(tick_marks, classes)
-
-# Floats to be round up to two decimal places if using normalize = True
-# or else use integers
- fmt ='.2f'if normalize else'd'
-# Sets threshold of 0.5
- thresh = cm.max() /2.
-# Iterate through the results and differentiate between two text colours
-# by using the threshold as a cut-off
-for i, j in itertools.product(range(cm.shape[0]), range(cm.shape[1])):
- plt.text(j, i, format(cm[i, j], fmt),
- horizontalalignment ="center",
- color ="white"if cm[i, j] > thresh else"black")
-
- plt.tight_layout()
- plt.ylabel('True label')
- plt.xlabel('Predicted label')
-
-
-
# Compute confusion matrix
-matrix = confusion_matrix(y_test, y_mp, labels = [0,1])
-np.set_printoptions(precision =2)
-
-# Plot confusion matrix without normalisation
-plt.figure()
-plot_confusion_matrix(matrix,
-# Define classes of outcomes
- classes = ['Max_Phase = 0','Max_Phase = 1'],
-# Set normalize = True if wanting ratios instead
- normalize =False,
- title ="Confusion matrix without normalisation"
- )
-
-
Confusion matrix, without normalization
-[[143 59]
- [ 54 123]]
-
-
-
-
-
-
A common rule of thumb for confusion matrix was that all predicted outcomes were columns and all the true outcomes were rows. However, there might be exceptions where this was the other way round. Four different categories could be seen in the confusion matrix which were:
-
-
True positive - Predicted Max_Phase = 1 & True Max_Phase = 1 (126 out of 189 samples)
-
True negative - Predicted Max_Phase = 0 & True Max_Phase = 0 (139 out of 190 samples)
-
False positive - Predicted Max_Phase = 1 & True Max_Phase = 0 (63 out of 189 samples)
-
False negative - Predicted Max_Phase = 0 & True Max_Phase = 1 (51 out of 190 samples)
-
-
By having these four categories known would then lead us to the next section about classification report, which showed all the precision, recall, f1-score and support metrics to evaluate the performance of this classifier.
-
-
-
-
Classification report
-
-
from sklearn.metrics import classification_report
-print(classification_report(y_test, y_mp))
Precision was a measure of the accuracy of a predicted outcome, where a class label had been predicted by the classifier. So in this case, we could see that for class label 1, the precision was 0.67, which corresponded to the true positive result of 126 out of 189 samples (= 0.666). It was defined by:
-
-
Recall, also known as sensitivity (especially widely used in biostatistics and medical diagnostic fields), was a measure of the strength of the classifier to predict a positive outcome. In simple words, it measured the true positive rate. In this example, there was a total of 126 out of 177 samples (which = 0.712, for True Max_Phase = 1 row) that had a true positive outcome of having a max phase of 1. It was defined by:
-
-
The precision and recall metrics could also be calculated for class label = 0, which were shown for the row 0 in the classification report.
-
f1-score, or also known as balanced F-score or F-measure, denoted the harmonic average of both precision and recall metrics. This metric would also give another indication about whether this model performed well on outcome predictions. It normally ranged from 0 (worst precision and recall) to 1 (perfect precision and recall). For this particular classifier, f1-score was at 0.69 (for class label = 1), which was definitely not at its worst, but also could be further improved. It was defined as:
-
-
Support, which some readers might have already worked out how the numbers were derived, was the total number of true samples in each class label (reading row-wise from the confusion matrix). The main purpose of showing this metric was to help clarifying whether the model or classifier had a reasonably balanced dataset for each class or otherwise.
-
-
-
-
Log loss
-
Log loss could be used as another gauge to show how good the classifier was at making the outcome predictions. The further the predicted probability was from the true value, the larger the log loss, which was also ranged from 0 to 1. Ideally, the smaller the log loss the better the model would be. Here, we had a log loss of 0.607 for this particular model.
-
-
# Log loss
-from sklearn.metrics import log_loss
-log_loss(y_test, y_mp_proba)
-
-
0.6171675830248284
-
-
-
-
-
-
Discussions and conclusion
-
So here I’ve completed a very basic LR classifier model for ChEMBL compound dataset. By no means was this a perfect ML model as I haven’t actually changed the default settings of scikit-learn’s LogisticRegression() classifier, with examples such as adjusting C, a regularization parameter which was set at ‘1.0’ by default, and also solvers, which could take in different algorithms for use in optimisation problems and normally set as ‘lbfgs’ by default.
-
So with this default LR model, the evaluation metrics demonstrated a LR classifer of moderate quality to predict the approval outcomes on ChEMBL small molecules, with a lot of rooms for improvements. Therefore, I could not yet confirm fully that the physicochemical parameters chosen would be the best ones to predict the approval outcomes for any small molecules. However, I might be okay to say that these molecular parameters were on the right track to help with making this prediction.
-
To further improve this model, I could possibly trial changing the C value and use different solvers to see if better outcomes could be achieved, or even add more molecular parameters in the model to test. I could have also trialled adding more class labels, e.g. making it between max phase 1, 2 and 4, or a mix-and-match between each max phase category. Other things to consider would be to use other types of ML methods such as naive Bayes, K-nearest neighbours or decision trees and so on. To tackle the problem thoroughly, I would most likely need to do an ensemble of different ML models to find out which model would be the most optimal to answer our target question.
-
-
-
-
Final words
-
I’ve experienced the fun of ML after completing this project. The idea was to build on what I knew gradually and enjoy what ML could do when making critical decisions. From what I’ve learnt about ML so far (and definitely more to learn) was that the quality of data was vital for making meaningful interpretations of the results.
-
However, jumping back to present time, I’ll need to work on my second project first, which is about using Rust interactively via Jupyter notebook. At the moment, I’m not sure how long it will take or how the content will play out. I’ll certainly do as much as I can since Rust is very new to me. If I get very stuck, I’d most likely continue on this ML series. Thanks for reading.
-
-
-
-
References
-
I’ve listed below most of the references used throughout this project. Again, huge thanks could not be forgotten for our online communities, and definitely also towards the references I’ve used here.
In a very rough sense, three main areas have been looked at (not exhaustive) with the aim to create safer therapeutic drugs:
-
-
Structural alerts on compound substructures that are known to cause adverse drug effects or pan-assay interference compounds (PAINs)
-
Many have looked into structural alerts (an example repo: rd_filters). ChEMBL database has already had a cheminformatic utils web service developed that provides structural alert computations for compounds. There are most likely much more efforts than these ones.
-
Toxicophores in relation to human ether-a-go-go-related gene (hERG) potassium channel (related to structural alerts as well)
-
hERG potassium channel is also another frequently-looked-at aspect for drug toxicology due to its known effect leading to cardiac QT prolongations or more commonly known as arrhythmias (Curran et al. 1995).
-
CYP enzymes with the well-known ones as CYP3A4, 2D6, 1A2, 2C9 and 2C19
-
CYP450 enzymes play a key role in the metabolism and toxicology parts of the ADMET process of drugs. When a drug behaves like a cytochrome inhibitor, it inhibits the activity of a particular cytochrome enzyme e.g. CYP3A4 leading to a reduction of clearance of a particular therapeutic drug e.g. a CYP3A4 substrate such as apixaban, thus increasing its plasma concentration in vivo causing a higher chance of adverse effect (which in the context of apixaban, this means the poor person taking the apixaban may get excessive bleeding…).
-
-
Other useful categories involve drug-induced skin sensitisations and liver injuries and more.
-
My very inital naive thought is that if we can at least cover some of the drug toxicology part during drug design and discovery process, this may be able to save some resources along the way (obviously it won’t be this simple…). The main thing here is that it may still be useful and interesting to look into the relationship between CYP450 and small drug molecules - to see if there are anything worth further explorations. This post will start with the two largest groups of CYP inhibitors, so focussing on CYP3A4 and 2D6 first.
-
While my focus is only on a very small cohort of small molecules relating to only two CYPs, it is also worth noting that there are actually more CYPs present as well, for example, CYP1A1, 2A6, 2B6, 2C8, 2E1, 2J2, 3A5 (note: amlodipine is a moderate CYP3A5 inhibitor and will be looked at below), 3A7 and 4F2 (Guengerich 2020). The cited paper here also provides quite a comprehensive background on the history of CYP450 and their relevance to toxicities in drugs, so I won’t repeat them here.
-
-
-
More on structural alerts
-
I am only really curious about the data sources used to build these ChEMBL structural alerts, so below are some of my notes on these sources.
-
From ChEMBL 20, only 6 filters are present, as shown by this ChEMBL blogpost - it may appear that this blog post cites all 8 filters but in fact it only has 6. I’ve attempted to find out the sources of these ChEMBL structural alert sets, here they are:
From ChEMBL 20 to 23, there are 8 filters in total (agreed with rd_filters’ README.md that there aren’t many documentations about this in ChEMBL, as I’ve tried also), the sources of the two additional ones are as follow:
-
-
Inpharmatica - unable to find direct source initially but this is later confirmed as private communications between ChEMBL and Inpharmatica Ltd. in the earlier days - an older ChEMBL presentation on ChEMBL 09 mentions about this, and this is also further elaborated by this paper (Gaulton et al. 2016)
-
SureChEMBL (old link provided by the paper (Gaulton et al. 2016) also no longer exists)
-
-
RDKit (section on “Filtering unwanted substructures”) also has another NIH filter based on two other references (Jadhav et al. 2010) and (Doveston et al. 2015). At one point I’m so confused with this NIH filter here and the NIH MLSMR one above… they are actually different as different papers are cited.
-
RDKit also uses the above 8 filters mentioned in ChEMBL in its FilterCatalogs class currently. Brenk filter seems to be the same as the CHEMBL_Dundee one since both of them have quoted the same journal paper as reference. It’s also got a ZINC one I think. Before I get very carried away, I’ll stop searching for every structural alerts papers here as there are many in the literatures.
-
-
-
-
More on CYPs and ADMET
-
A bit of a sidetrack for this part (feel free to skip) as I come across a new paper online recently about using deep learning model for ADMET prediction which uses data from Therapeutics data commons (TDC). So while working on this relevant topic of CYP and ADMET (only the metabolism and toxicology parts), I just want to dig a bit deeper to see what sort of data are used by TDC.
-
The TDC ADME dataset, specifically the metabolism one on all five CYP isoenzymes (CYP2C19, 2D6, 3A4, 1A2 and 2C9), are all derived from a 2009 paper by Veith et al.. A closer look at this paper only seems to mention:
-
-
…we tested 17,143 samples at between seven and fifteen concentrations for all five CYP isozymes. The samples consisted of 8,019 compounds from the MLSMR including compounds chosen for diversity and rule-of-five compliance, 16 synthetic tractability, and availability; 6,144 compounds from a set of biofocused libraries which included 1,114 FDA-approved drugs; and 2,980 compounds from combinatorial libraries containing privileged structures targeted at GPCRs and kinases, and libraries of purified natural products or related structures…
-
-
If I go to its original journal paper site (the link provided was a NCBI one), there is only one additional Excel file with a long list of chemical scaffolds showing different CYP activities (no other supplementary information I can spot there). The only likely lists of compounds tested are shown in its figures 6 and 7 in the paper, where figure 7 is more relevant for drug-drug interactions. I then realise the proportions of FDA-approved drugs used and the rest of the molecules tested in this paper are also not very balanced (thinking along the line of approved drugs and non-approved drugs), and notice that what they are saying in its discussion about how they are not noticing the usual prominent activities of CYP3A4 and 2D6 in the compounds they’ve tested:
-
-
…It has been suggested that CYP 3A4 is the most prominent P450 isozyme in drug metabolism and hepatic distribution (Fig. 2b),25,26 but the drugs in our collection do not appear to have been optimized away from this activity. There has also been speculation that CYP 2D6 isozyme plays a prominent role in drug metabolism,27 but no difference in activity was observed between diversity compounds and approved drugs for this isozyme…
-
-
I wonder if this may be due to the imbalanced set of compounds used e.g. number of FDA-approved drug (smaller) vs. number of other compounds from other libraries (larger)…
-
I’ve also visited FDA’s website to look at how the CYP stories are compiled (FDA link). The in vitro inhibitors and clinical index inhibitors are not completely the same across all the CYPs. There are some overlappings in CYP3A4/5 and 2D6 for sure but definitely not exactly the same across all the documented CYPs in this FDA webpage.
-
So back to this new paper on predicting ADMET… how likely will it be useful in real-life hit/lead ADMET optimisation projects in drug discovery settings if the data source only involves a larger portion of non-approved drugs versus a smaller portion of actual FDA-approved drugs?… It just shows that there are a lot of things to think about in the DMPK/ADMET areas within drug discovery pipelines, as ultimately this is crucial to see if a candidate molecule will proceed or not (i.e. causing toxicity or not and whether it’s tolerable side effects or adverse or even life-threatening ones instead).
Some of the code blocks have been folded to keep the post length a bit more manageable - click on the code links to see full code (only applies to the HTML version, not the Jupyter notebook version).
-
-
-
-
Extracting data
-
First step here is to import the following software packages in order to retrieve and work with ChEMBL data (again).
+
+
Import libraries
+
+Code
import pandas as pd
-import chembl_downloader
-from chembl_downloader import latest
-from rdkit import Chem
-from rdkit.Chem import Draw, AllChem
-# For maximum common substructures & labelling stereocentres
-from rdkit.Chem import rdFMCS, rdCIPLabeler
-from rdkit.Chem.Draw import IPythonConsole
-IPythonConsole.drawOptions.addAtomIndices =False
-# Change to false to remove stereochem labels
-IPythonConsole.drawOptions.addStereoAnnotation =True
-IPythonConsole.ipython_useSVG=True
+import torch
+import torch.nn as nn
+import torch.nn.functional as F
+from torch.nn.functional import one_hot
+from torch.utils.data import TensorDataset, DataLoader
+import numpy as np
+import datamol as dm
+import rdkit
+from rdkit import Chem
+from rdkit.Chem import rdFingerprintGenerator
+import useful_rdkit_utils as uru
+import sys
+from matplotlib import pyplot as plt
+print(f"Pandas version used: {pd.__version__}")
+print(f"PyTorch version used: {torch.__version__}")
+print(f"NumPy version used: {np.__version__}")
+print(f"RDKit version used: {rdkit.__version__}")
+print(f"Python version used: {sys.version}")
+
+
+
Pandas version used: 2.2.3
+PyTorch version used: 2.2.2
+NumPy version used: 1.26.4
+RDKit version used: 2024.09.4
+Python version used: 3.12.7 (v3.12.7:0b05ead877f, Sep 30 2024, 23:18:00) [Clang 13.0.0 (clang-1300.0.29.30)]
+
+
+
+
+
Import adverse drug reactions (ADRs) data
+
This is an extremely small set of data compiled manually (by me) via references stated in the dataframe. For details about what and how the data are collected, I’ve prepared a separate post as a data note (add post link) to explain key things about the data. It may not lead to a very significant result but it is done as an example of what an early or basic deep neural network (DNN) model may look like. Ideally there should be more training data and also more features added or used. I’ve hypothetically set the goal of this introductory piece to predict therapeutic drug classes from ADRs, molecular fingerprints and cytochrome P450 substrate strengths, but this won’t be achieved in this initial post (yet).
-
# Latest version of ChEMBL
-latest_version = latest()
-print(f"The latest ChEMBL version is: {latest_version}")
+
data = pd.read_csv("All_CYP3A4_substrates")
+print(data.shape)
+data.head(3)
-
The latest ChEMBL version is: 34
-
+
(27, 8)
-
I’m using SQL via chembl_downloader to download approved drugs with their ChEMBL ID and equivalent canonical SMILES. All of the CYP3A4 and 2D6 inhibitors extracted from ChEMBL are based on the Flockhart table of drug interactions(Flockhart et al. 2021).
-
Note: Three other categories of medicines are not going to be looked at for now, which are the weak inhibitors, ones with in vitro evidence only and ones that are still pending reviews.
-
A bit about retrieving data here, the following may not be the best way to get the data, but I’ve somehow incorporated chembl_downloader into my own small piece of function code (see Python script named as “cyp_drugs.py” in the repo) to retrieve SMILES of approved drugs (other public databases may also work very well equally, but I’m used to using ChEMBL now as it’s easy to read and navigate).
-
Another possible way is to use get_target_sql() within chembl_downloader, e.g. using a specific CYP enzyme as the protein target to retrieve data, but it appears that there are no clear data marked to indicate the potency of CYP inhibition or induction (i.e. weak, moderate or strong) in the ChEMBL database (an example link for CYP2D6 in ChEMBL). The Flockhart table has clearly annotated each approved drug with journal paper citations so I decide to stick with the previous method.
-
-
## Main issue previously is with sql string - too many quotation marks!
-# e.g. WHERE molecule_dictionary.pref_name = '('KETOCONAZOLE', 'FLUCONAZOLE')'': near "KETOCONAZOLE": syntax error
-# Resolved issue by adding string methods e.g. strip() and replace() to sql query string
-
-from cyp_drugs import chembl_drugs
-
-# Get a list of strong cyp3a4 inhibitors
-# For the story on why I also added a weird spelling of "itraconzole", please see below.
-# and save as a tsv file
-df_3a4_strong_inh = chembl_drugs(
-"CERITINIB", "CLARITHROMYCIN", "DELAVIRIDINE", "IDELALISIB", "INDINAVIR", "ITRACONAZOLE", "ITRACONZOLE", "KETOCONAZOLE", "MIBEFRADIL", "NEFAZODONE", "NELFINAVIR", "RIBOCICLIB", "RITONAVIR", "SAQUINAVIR", "TELAPREVIR", "TELITHROMYCIN", "TUCATINIB", "VORICONAZOLE",
-#file_name="strong_3a4_inh"
- )
-df_3a4_strong_inh.head()
-
+
@@ -157,62 +76,69 @@
-
chembl_id
-
pref_name
-
max_phase
-
canonical_smiles
+
generic_drug_name
+
notes
+
cyp_strength_of_evidence
+
drug_class
+
adverse_drug_reactions
+
first_ref
+
second_ref
+
date_checked
0
-
CHEMBL2403108
-
CERITINIB
-
4.0
-
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
+
carbamazepine
+
NaN
+
strong
+
antiepileptics
+
constipation^^, leucopenia^^, dizziness^^, som...
+
drugs.com
+
nzf
+
211024
1
-
CHEMBL1741
-
CLARITHROMYCIN
-
4.0
-
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
+
eliglustat
+
NaN
+
strong
+
metabolic_agents
+
diarrhea^^, oropharyngeal_pain^^, arthralgia^^...
+
drugs.com
+
emc
+
151124
2
-
CHEMBL2216870
-
IDELALISIB
-
4.0
-
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
-
-
-
3
-
CHEMBL115
-
INDINAVIR
-
4.0
-
CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](...
-
-
-
4
-
CHEMBL22587
-
ITRACONAZOLE
+
flibanserin
NaN
-
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5CO[C@](...
+
strong
+
CNS_agents
+
dizziness^^, somnolence^^, sedation^, fatigue^...
+
drugs.com
+
Drugs@FDA
+
161124
-
-
## Get a list of moderate cyp3a4 inhibitors
-# skipping grapefruit juice as it's not quite an approved drug...
-# note: amlodipine inhibits cyp3a5
-df_3a4_mod_inh = chembl_drugs(
-"AMLODIPINE", "APREPITANT", "CIPROFLOXACIN", "CRIZOTINIB", "DILTIAZEM", "ERYTHROMYCIN", "FLUCONAZOLE", "IMATINIB", "LETERMOVIR", "NETUPITANT", "VERAPAMIL", #file_name="mod_3a4_inh"
- )
-df_3a4_mod_inh.head()
-
+
For drug with astericks marked in “notes” column, see data notes under “Exceptions for ADRs” section in 1_ADR_data.qmd (separate post).
+
I’m dropping some of the columns that are not going to be used later.
Before extracting data from ChEMBL, I’m getting a list of drug names in capital letters ready first which can be fed into chembl_downloader with my old cyp_drugs.py to retrieve the SMILES of these drugs.
+
+
+Code
+
string = df["generic_drug_name"].tolist()
+# Convert list of drugs into multiple strings of drug names
+drugs =f"'{"','".join(string)}'"
+# Convert from lower case to upper case
+for letter in drugs:
+if letter.islower():
+ drugs = drugs.replace(letter, letter.upper())
+print(drugs)
# Get a list of strong cyp2d6 inhibitors
-df_2d6_strong_inh = chembl_drugs(
-"BUPROPION", "FLUOXETINE", "PAROXETINE", "QUINIDINE",
-#file_name="strong_2d6_inh"
- )
-df_2d6_strong_inh
+
+Code
+
# Get SMILES for each drug (via copying-and-pasting the previous cell output - attempted various ways to feed the string
+# directly into cyp_drugs.py, current way seems to be the most straightforward one...)
+from cyp_drugs import chembl_drugs
+# Using ChEMBL version 34
+df_3a4 = chembl_drugs(
+'CARBAMAZEPINE','ELIGLUSTAT','FLIBANSERIN','IMATINIB','IBRUTINIB','NERATINIB','ESOMEPRAZOLE','OMEPRAZOLE','IVACAFTOR','NALOXEGOL','OXYCODONE','SIROLIMUS','TERFENADINE','DIAZEPAM','HYDROCORTISONE','LANSOPRAZOLE','PANTOPRAZOLE','LERCANIDIPINE','NALDEMEDINE','NELFINAVIR','TELAPREVIR','ONDANSETRON','QUININE','RIBOCICLIB','SUVOREXANT','TELITHROMYCIN','TEMSIROLIMUS',
+#file_name="All_cyp3a4_smiles"
+ )
+print(df_3a4.shape)
+df_3a4.head(3)
+
+## Note: latest ChEMBL version 35 (as from 1st Dec 2024) seems to be taking a long time to load (no output after ~7min),
+## both versions 33 & 34 are ok with outputs loading within a few secs
+
+
+
(27, 4)
+
@@ -291,51 +237,45 @@
0
-
CHEMBL894
-
BUPROPION
-
4.0
-
CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1
+
CHEMBL108
+
CARBAMAZEPINE
+
4
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
1
-
CHEMBL41
-
FLUOXETINE
-
4.0
-
CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1
+
CHEMBL12
+
DIAZEPAM
+
4
+
CN1C(=O)CN=C(c2ccccc2)c2cc(Cl)ccc21
2
-
CHEMBL490
-
PAROXETINE
-
4.0
-
Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1
-
-
-
3
-
CHEMBL21578
-
QUINIDINE
-
NaN
-
C=C[C@H]1CN2CCC1C[C@@H]2[C@@H](O)c1ccnc2ccc(OC...
-
-
-
4
-
CHEMBL1294
-
QUINIDINE
-
4.0
-
C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc...
+
CHEMBL2110588
+
ELIGLUSTAT
+
4
+
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...
+
+
+
+
Merge dataframes
+
Next, I’m renaming the drug name column and merging the two dataframes together where one contains the ADRs and the other one contains the SMILES. I’m also making sure all drug names are in upper case for both dataframes so they can merge properly.
-
# Get a list of moderate cyp2d6 inhibitors
-df_2d6_mod_inh = chembl_drugs(
-"ABIRATERONE", "CINACALCET", "CLOBAZAM", "DOXEPIN", "DULOXETINE", "HALOFANTRINE", "LORCASERIN", "MOCLOBEMIDE", "ROLAPITANT", "TERBINAFINE",
-#file_name="mod_2d6_inh"
- )
-df_2d6_mod_inh.head()
Initially, four categories of approved drugs are retrieved - the strong and moderate CYP3A4 inhibitors, and also the strong and moderate CYP2D6 inhibitors. CYP3A4 inhibitors are the largest cohort of all the cytochrome inhibitors known so far (based on clinical documentations).
-
-
Import and preprocess data
+
+
Parse SMILES
+
Then I’m parsing the canonical SMILES through my old script to generate these small molecules as RDKit molecules and standardised SMILES, making sure these SMILES are parsable.
-
## When using pandas 2.2.2, numpy 2.0.0 and rdkit 2024.3.1
-# (all latest major versions at the time of writing,
-# note: rdkit has a latest minor release as 2024.03.4, which includes a patch for numpy 2.0)
-# Seems to work as a new df is generated but with error messages shown
-
-## Eventually using downgraded versions of pandas and numpy instead
-# pandas 2.1.4, numpy 1.26.4 & rdkit 2024.3.1 work with no error messages generated
-
-# preprocess canonical smiles
-from mol_prep import preprocess
-
-# cyp3a4 strong inhibitors
-df_3a4_s_inh = df_3a4_strong_inh.copy()
-df_3a4_s_inh_p = df_3a4_s_inh.apply(preprocess, axis=1)
-df_3a4_s_inh_p.head(3)
+
+Code
+
# Using my previous code to preprocess small mols
+# disable rdkit messages
+dm.disable_rdkit_log()
+
+# The following function code were adapted from datamol.io
+def preprocess(row):
+
+"""
+ Function to preprocess, fix, standardise, sanitise compounds
+ and then generate various molecular representations based on these molecules.
+ Can be utilised as df.apply(preprocess, axis=1).
+
+ :param smiles_column: SMILES column name (needs to be names as "canonical_smiles")
+ derived from ChEMBL database (or any other sources) via an input dataframe
+ :param mol: RDKit molecules
+ :return: preprocessed RDKit molecules, standardised SMILES, SELFIES,
+ InChI and InChI keys added as separate columns in the dataframe
+ """
+
+# smiles_column = strings object
+ smiles_column ="canonical_smiles"
+# Convert each compound into a RDKit molecule in the smiles column
+ mol = dm.to_mol(row[smiles_column], ordered=True)
+# Fix common errors in the molecules
+ mol = dm.fix_mol(mol)
+# Sanitise the molecules
+ mol = dm.sanitize_mol(mol, sanifix=True, charge_neutral=False)
+# Standardise the molecules
+ mol = dm.standardize_mol(
+ mol,
+# Switch on to disconnect metal ions
+ disconnect_metals=True,
+ normalize=True,
+ reionize=True,
+# Switch on "uncharge" to neutralise charges
+ uncharge=True,
+# Taking care of stereochemistries of compounds
+# Note: this uses the older approach of "AssignStereochemistry()" from RDKit
+# https://github.com/datamol-io/datamol/blob/main/datamol/mol.py#L488
+ stereo=True,
+ )
+
+# Adding following rows of different molecular representations
+ row["rdkit_mol"] = dm.to_mol(mol)
+ row["standard_smiles"] = dm.standardize_smiles(str(dm.to_smiles(mol)))
+#row["selfies"] = dm.to_selfies(mol)
+#row["inchi"] = dm.to_inchi(mol)
+#row["inchikey"] = dm.to_inchikey(mol)
+return row
+
+df_p3a4 = df.apply(preprocess, axis =1)
+print(df_p3a4.shape)
+df_p3a4.head(3)
+
+
+
(27, 9)
+
@@ -420,65 +402,107 @@
-
chembl_id
pref_name
+
cyp_strength_of_evidence
+
drug_class
+
adverse_drug_reactions
+
chembl_id
max_phase
canonical_smiles
rdkit_mol
standard_smiles
-
selfies
-
inchi
-
inchikey
0
-
CHEMBL2403108
-
CERITINIB
-
4.0
-
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
-
<rdkit.Chem.rdchem.Mol object at 0x135da19a0>
-
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
-
[C][C][=C][C][Branch2][Ring2][=Branch1][N][C][...
-
InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-...
-
VERWOWGGCGHDQE-UHFFFAOYSA-N
+
CARBAMAZEPINE
+
strong
+
antiepileptics
+
constipation^^, leucopenia^^, dizziness^^, som...
+
CHEMBL108
+
4
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2dee0>
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
1
-
CHEMBL1741
-
CLARITHROMYCIN
-
4.0
-
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1b60>
-
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
-
[C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran...
-
InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21...
-
AGOYDEPGAOXOCK-KCBOHYOISA-N
+
ELIGLUSTAT
+
strong
+
metabolic_agents
+
diarrhea^^, oropharyngeal_pain^^, arthralgia^^...
+
CHEMBL2110588
+
4
+
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2dfc0>
+
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...
2
-
CHEMBL2216870
-
IDELALISIB
-
4.0
-
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1a80>
-
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
-
[C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][...
-
InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-...
-
IFSDAJWBUCMOAH-HNNXBMFYSA-N
+
FLIBANSERIN
+
strong
+
CNS_agents
+
dizziness^^, somnolence^^, sedation^, fatigue^...
+
CHEMBL231068
+
4
+
O=c1[nH]c2ccccc2n1CCN1CCN(c2cccc(C(F)(F)F)c2)CC1
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2e030>
+
O=c1[nH]c2ccccc2n1CCN1CCN(c2cccc(C(F)(F)F)c2)CC1
+
+
+
+
Split data
+
Random splits usually lead to overly optimistic models, where testing molecules are too similar to traininig molecules leading to many problems. This is further discussed in two other blog posts that I’ve found useful - post by Greg Landrum and post by Pat Walters.
+
Here I’m trying out Pat’s useful_rdkit_utils’ GroupKFoldShuffle code (code originated from this thread) to split data (Butina clustering/splits). To do this, it requires SMILES to generate molecular fingerprints which will be used in the training and testing sets (potentially for future posts and in real-life cases, more things can be done with the SMILES or other molecular representations for machine learning, but to keep this post easy-to-read, I’ll stick with only generating the Morgan fingerprints for now).
# Generate numpy arrays containing the fingerprints
+df_p3a4['fp'] = df_p3a4.rdkit_mol.apply(rdFingerprintGenerator.GetMorganGenerator().GetCountFingerprintAsNumPy)
+
+# Get Butina cluster labels
+df_p3a4["butina_cluster"] = uru.get_butina_clusters(df_p3a4.standard_smiles)
+
+# Set up a GroupKFoldShuffle object
+group_kfold_shuffle = uru.GroupKFoldShuffle(n_splits=5, shuffle=True)
+
+# Using cross-validation/doing data split
+## X = np.stack(df_s3a4.fp), y = df.adverse_drug_reactions, group labels = df_s3a4.butina_cluster
+for train, test in group_kfold_shuffle.split(np.stack(df_p3a4.fp), df.adverse_drug_reactions, df_p3a4.butina_cluster):
+print(len(train),len(test))
+
+
+
17 10
+23 4
+23 4
+23 4
+22 5
+
+
+
+
+
+
Locate training and testing sets after data split
+
While trying to figure out how to locate training and testing sets after the data split, I’ve gone into a mini rabbit hole myself (a self-confusing session but gladly it clears up when my thought process goes further…). For example, some of the ways I’ve planned to try: create a dictionary as {index: butina label} first - butina cluster labels vs. index e.g. df_s3a4[“butina_cluster”], or maybe can directly convert from NumPy array to tensor - will need to locate drugs via indices first to specify training and testing sets, e.g. torch_train = torch.from_numpy(train) or torch_test = torch.from_numpy(test). It is actually simpler than this, which is to use pd.DataFrame.iloc() as shown below.
# Convert indices into list
+train_set = train.tolist()
+# Locate drugs and drug info via pd.DataFrame.iloc
+df_train = df_p3a4.iloc[train_set]
+print(df_train.shape)
+df_train.head(2)
Here what I’m trying to do is to check structural validities of all the drug molecules, and one of the easiest things to do is to look at their chemical structures directly.
Set up training and testing sets for X and y variables
+
This part involves converting X (features) and y (target) variables into either one-hot encodings or vector embeddings, since I’ll be dealing with categories/words/ADRs and not numbers, and also to split each X and y variables into training and testing sets. At the very beginning, I’ve thought about using scikit_learn’s train_test_split(), but then realised that I should not need to do this as it’s already been done in the previous step (obviously I’m confusing myself again…). Essentially, this step can be integrated with the one-hot encoding and vector embeddings part as shown below.
+
There are three coding issues that have triggered warning messages when I’m trying to figure out how to convert CYP strengths into one-hot encodings:
+
+
A useful thread has helped me to solve the downcasting issue in pd.DataFrame.replace() when trying to do one-hot encoding to replace the CYP strengths for each drug
+
A Pandas setting-with-copy warning shows if using df[“column_name”]:
+
+
+
A value is trying to be set on a copy of a slice from a DataFrame. Try using .loc[row_indexer,col_indexer] = value instead
+
+
The solution is to enable the copy-on-write globally (as commented in the code below; from Pandas reference).
+
+
PyTorch user warning appers if using df_train[“cyp_strength_of_evidence”].values, as this leads to non-writable tensors with a warning like this:
+
+
+
UserWarning: The given NumPy array is not writable, and PyTorch does not support non-writable tensors. This means writing to this tensor will result in undefined behavior. You may want to copy the array to protect its data or make it writable before converting it to a tensor. This type of warning will be suppressed for the rest of this program. (Triggered internally at /Users/runner/work/pytorch/pytorch/pytorch/torch/csrc/utils/tensor_numpy.cpp:212.)
+
+
One of the solutions is to add copy() e.g. col_encoded = one_hot(torch.from_numpy(df[“column_name”].values.copy()) % total_numbers_in_column) or alternatively, convert column into numpy array first, then make the numpy array writeable (which is what I’ve used in the code below).
+
+
+Code
+
## X_train
+# 1. Convert "cyp_strength_of_evidence" column into one-hot encoding
+# Enable copy-on-write globally to remove the warning
+pd.options.mode.copy_on_write =True
+
+# Replace CYP strength as numbers
+with pd.option_context('future.no_silent_downcasting', True):
+ df_train["cyp_strength_of_evidence"] = df_train["cyp_strength_of_evidence"].replace({"strong": 1, "mod": 2}).infer_objects()
+ df_test["cyp_strength_of_evidence"] = df_test["cyp_strength_of_evidence"].replace({"strong": 1, "mod": 2}).infer_objects()
+
+# Get total number of CYP strengths in df
+total_cyp_str_train =len(set(df_train["cyp_strength_of_evidence"]))
+
+# Convert column into numpy array first, then make the numpy array writeable
+cyp_array_train = df_train["cyp_strength_of_evidence"].to_numpy()
+cyp_array_train.flags.writeable =True
+cyp_str_train_t = one_hot(torch.from_numpy(cyp_array_train) % total_cyp_str_train)
+cyp_str_train_t
Without going into too much details about vector embeddings (as there are a lot of useful learning materials about it online and in texts), here’s roughly how I understand embeddings while working on this post. Embeddings are real-valued dense vectors that are normally in multi-dimensional arrays and they can represent and catch the context of a word or sentence, the semantic similarity and especially the relation of each word with other words in a corpus of texts. They roughly form the basis of natural language processing and also contribute to how large language models are built… in a very simplified sense, but obviously this can get complex if we want the models to do more. Here, I’m trying something experimental so I’m going to convert each ADR for each drug into embeddings.
+
+
+Code
+
# 2. Convert "adverse_drug_reactions" column into embeddings
+## see separate scripts used previously e.g. words_tensors.py
+## or Tensors_for_adrs_interactive.py to show step-by-step conversions from words to tensors
+
+# Save all ADRs from common ADRs column as a list (joining every row of ADRs in place only)
+adr_str_train = df_train["adverse_drug_reactions"].tolist()
+# Join separate rows of strings into one complete string
+adr_string_train =",".join(adr_str_train)
+# Converting all ADRs into Torch tensors using words_tensors.py
+from words_tensors import words_tensors
+adr_train_t = words_tensors(adr_string_train)
+adr_train_t
When trying to convert the “fp” column into tensors, there is one coding issue I’ve found relating to the data split step earlier. Each time the notebook is re-run with the kernel refreshed, the data split will lead to different proportions of training and testing sets due to the “shuffle = True”, which subsequently leads to different training and testing set arrays. One of the ways to circumvent this is to turn off the shuffle but this is not ideal for model training. So an alternative way that I’ve tried is to use ndarray.size (which is the product of elements in ndarray.shape, equivalent to multiplying the numbers of rows and columns), and divide the row of the intended tensor shape by 2 as I’m trying to reshape training arrays so they’re all in 2 columns in order for torch.cat() to work later.
+
+
+Code
+
# 3. Convert "fp" column into tensors
+# Stack numpy arrays in fingerprint column
+fp_train_array = np.stack(df_train["fp"])
+# Convert numpy array data type from uint32 to int32
+fp_train_array = fp_train_array.astype("int32")
+# Create tensors from array
+fp_train_t = torch.from_numpy(fp_train_array)
+# Reshape tensors
+fp_train_t = torch.reshape(fp_train_t, (int(fp_train_array.size/2), 2))
+fp_train_t.shape # tensor.ndim to check tensor dimensions
+
+
+
torch.Size([22528, 2])
+
+
+
+
adr_train_t.shape
+
+
torch.Size([674, 2])
+
+
+
+
cyp_str_train_t.shape
+
+
torch.Size([22, 2])
+
+
+
+
# Concatenate adr tensors, fingerprint tensors and cyp strength tensors as X_train
+X_train = torch.cat([adr_train_t, fp_train_t, cyp_str_train_t], 0).float()
+X_train
## y_train
+# Use drug_class column as target
+# Convert "drug_class" column into embeddings
+# total number of drug classes in df = 20 - len(set(df["drug_class"])) - using embeddings instead of one-hot
+dc_str_train = df_train["drug_class"].tolist()
+dc_string_train =",".join(dc_str_train)
+y_train = words_tensors(dc_string_train)
+y_train
This is a small detour while checking structural validities due to the presence of two duplicated molecules, and since these two molecules consist of stereocentres, I’m just going to have a look at their stereochemistries.
+
+
+
Input preprocessing pipeline using PyTorch Dataset and DataLoader
+
There is a size-mismatch-between-tensors warning when I’m trying to use PyTorch’s TensorDataset(). I’ve found out that to use the data loader and tensor dataset, the first dimension of all tensors needs to be the same. Initially, they’re not, where X_train.shape = [24313, 2], y_train.shape = [1, 2]. Eventually I’ve settled on two ways that can help with this:
+
+
use tensor.unsqueeze(dim = 1) or
+
use tensor[None] which’ll insert a new dimension at the beginning, then it becomes: X_train.shape = [1, 24313, 2], y_train.shape = [1, 1, 2]
+
+
+
X_train[None].shape
+
+
torch.Size([1, 23224, 2])
+
+
+
+
X_train.shape
+
+
torch.Size([23224, 2])
+
+
+
+
y_train[None].shape
+
+
torch.Size([1, 1, 2])
+
+
+
+
y_train.shape
+
+
torch.Size([1, 2])
+
+
+
+
# Create a PyTorch dataset on training data set
+train_data = TensorDataset(X_train[None], y_train[None])
+# Sets a seed number to generate random numbers
+torch.manual_seed(1)
+batch_size =1
+
+# Create a dataset loader
+train_dl = DataLoader(train_data, batch_size, shuffle =True)
+
+
+
# Create another PyTorch dataset on testing data set
+test_data = TensorDataset(X_test[None], y_test[None])
+torch.manual_seed(1)
+batch_size =1
+test_dl = DataLoader(test_data, batch_size, shuffle=True)
+
-
-
quinidine
-
There are different stereochemistries spotted in the two quinidines shown below.
-
-
# Stereochem in RDKit
-# Older approach - AssignStereochemistry() -> this is used in datamol's standardize_mol(),
-# which is used in my small mol_prep.py script
-# Newer approach - FindPotentialStereo()
+
+
Set up a simple DNN regression model
+
I’m only going to use a very simple two-layer DNN model to match the tiny dataset used here. There are many other types of neural network layers or bits and pieces that can be used to suit the goals and purposes of the dataset used. This reference link shows different types of neural network layers that can be used in PyTorch.
+
Below are some short notes regarding a neural network (NN) model:
+
+
goal of the model is to minimise loss function L(W) (where W = weight) to get the optimal model weights
+
matrix with W (for hidden layer) connects input to hidden layer; matrix with W (for outer layer) connects hidden to output layer
+
Input layer -> activation function of hidden layer -> hidden layer -> activation function of output layer -> output layer (a very-simplified flow diagram to show how the layers get connected to each other)
+
+
About backpropagation for loss function:
+
+
backpropagation is a computationally efficient way to calculate partial derivatives of loss function to update weights in multi-layer NNs
+
it’s based on calculus chain rule to compute derivatives of mathematical functions (automatic differentiation)
+
matrix-vector multiplications in backpropagation are computationally more efficient to calculate than matrix-matrix multiplications e.g. forward propagation
# note: this is a very simple two-layer NN model only
+
+# Set up hidden units between two connected layers - one layer with 6 hidden units and the other with 3 hidden units
+hidden_units = [6, 3]
+# Input size same as number of columns in X_train
+input_size = X_train.shape[1]
+# Initiate NN layers as a list
+all_layers = []
+
+## Specify how the input, hidden and output layers are going to be connected
+# For each hidden unit within the hidden units specified above:
+for h_unit in hidden_units:
+# specify sizes of input sample (input size = X_train col size) & output sample (hidden units) in each layer
+# https://pytorch.org/docs/stable/generated/torch.nn.Linear.html#torch.nn.Linear
+ layer = nn.Linear(input_size, h_unit)
+# add each layer
+ all_layers.append(layer)
+# add activation function (trying rectified linear unit) for next layer
+ all_layers.append(nn.ReLU())
+# for the next layer to be added, the input size will be the same size as the hidden unit
+ input_size = h_unit
+
+# Specify the last layer (where input_feature = hidden_units[-1] = 3)
+all_layers.append(nn.Linear(hidden_units[-1], 1))
+
+# Set up a container that'll connect all layers in the specified sequence in the model
+model = nn.Sequential(*all_layers)
+model
# Get 2D image of quinidine at row 3
-df_2d6_s_inh_p.loc[3, "rdkit_mol"]
-
-
-
+
+
+
+
Train model
+
This part is mainly about defining the loss function when training the model with the training data, and optimising model by using a stochastic gradient descent. One key thing I’ve gathered from trying to learn about deep learning is that we’re aiming for global minima and not local minima (e.g. if learning rate is too small, this may end up with local minima; if learning rate is too large, it may end up over-estimating the global minima). I’ve also encountered the PyTorch padding method to make sure the input and target tensors are of the same size, otherwise the model will run into matrix broadcasting issue (which will likely influence the results). The training loss appears to have converged when the epoch runs reach 100 and/or after (note this may vary due to shuffle data sampling)… (I also think my data size is way too small to show a clear contrast in training loss convergence).
+
References for: nn.MSELoss() - measures mean squared error between X and y, and nn.functional.pad() - pads tensor (increase tensor size)
+
Obtaining training loss via model training:
+
+
+Code
+
# Set up loss function
+loss_f = nn.MSELoss()
+# Set up stochastic gradient descent optimiser to optimise model (minimise loss) during training
+# lr = learning rate - default: 0.049787 (1*e^-3)
+optim = torch.optim.SGD(model.parameters(), lr=0.005)
+# Set training epochs (epoch: each cycle of training or passing through the training set)
+num_epochs =200
+# Set the log output to show training loss - for every 20 epochs
+log_epochs =20
+torch.manual_seed(1)
+# Create empty lists to save training loss (for training and testing/validation sets)
+train_epoch_loss = []
+test_epoch_loss = []
+
+# Predict via training X_batch & obtain train loss via loss function from X_batch & y_batch
+for epoch inrange(num_epochs):
+ train_loss =0
+for X_batch, y_batch in train_dl:
+# Make predictions
+ predict = model(X_batch)[:, 0]
+# Make input tensors the same size as y_batch tensors
+ predict_pad = F.pad(predict[None], pad=(1, 0, 0, 0))
+# Calculate training loss
+ loss = loss_f(predict_pad, y_batch)
+# Calculate gradients (backpropagations)
+ loss.backward(retain_graph=True)
+# Update parameters using gradients
+ optim.step()
+# Reset gradients back to zero
+ optim.zero_grad()
+ train_loss += loss.item()
+
+if epoch % log_epochs ==0:
+print(f"Epoch {epoch} Loss {train_loss/len(train_dl):.4f}")
+
+ train_epoch_loss.append(train_loss)
+
+
+
Epoch 0 Loss 4.3253
+Epoch 20 Loss 3.5549
+Epoch 40 Loss 2.9739
+Epoch 60 Loss 2.4838
+Epoch 80 Loss 2.1047
-
-
# Get 2D image of quinidine at row 4
-df_2d6_s_inh_p.loc[4, "rdkit_mol"]
-
-
-
+
+
Epoch 100 Loss 1.8545
+Epoch 120 Loss 1.7217
+Epoch 140 Loss 1.6662
+Epoch 160 Loss 1.6471
+Epoch 180 Loss 1.6415
-
-
# Get SMILES for quinidine at row 3
-df_2d6_s_inh_p.loc[3, "canonical_smiles"]
# Predict via testing X_batch & obtain test loss
+for epoch inrange(num_epochs):
+ test_loss =0
+for X_batch, y_batch in test_dl:
+# Make predictions
+ predict_test = model(X_batch)[:, 0]
+# Make input tensors the same size as y_batch tensors
+ predict_pad_test = F.pad(predict_test[None], pad=(1, 0, 0, 0))
+# Calculate training loss
+ loss = loss_f(predict_pad_test, y_batch)
+# Calculate gradients (backpropagations)
+ loss.backward(retain_graph=True)
+# Update parameters using gradients
+ optim.step()
+# Reset gradients back to zero
+ optim.zero_grad()
+ test_loss += loss.item()
+
+if epoch % log_epochs ==0:
+print(f"Epoch {epoch} Loss {test_loss/len(test_dl):.4f}")
+
+ test_epoch_loss.append(test_loss)
+
+
+
Epoch 0 Loss 0.4037
+Epoch 20 Loss 0.1767
+Epoch 40 Loss 0.0963
+Epoch 60 Loss 0.0615
+Epoch 80 Loss 0.0452
+Epoch 100 Loss 0.0373
+Epoch 120 Loss 0.0335
+
+
Epoch 140 Loss 0.0316
+Epoch 160 Loss 0.0306
+Epoch 180 Loss 0.0301
-
-
# Get SMILES for quinidine at row 4
-df_2d6_s_inh_p.loc[4, "canonical_smiles"]
# quinidine index row 4
-quinidine_4 = Chem.MolFromSmiles('C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2ccc(OC)cc12')
-rdCIPLabeler.AssignCIPLabels(quinidine_4)
-quinidine_4
-
-
-
+
At the moment, when this notebook is re-run on a refreshed kernel, this leads to a different train and test split each time, and also leading to a different train and test (validation) loss each time. There may be two types of scenarios shown in the plot above where:
+
+
test loss is higher than train loss (overfitting) - showing the model may be way too simplified and is likely under-trained
+
train loss is higher than test loss (underfitting) - showing that the model may not have been trained well, and is unable to learn the features in the training data and apply them to the test data
+
+
When there are actually more training data available with also other hyperparameters fine tuned, it may be possible to see another scenario where both test loss and train loss are very similar in trend, meaning the model is being trained well and able to generalise the training to the unseen data.
+
To mitigate overfitting:
+
+
firstly there should be more training data than what I’ve had here
+
use L1 or L2 regularisation to minimise model complexity by adding penalities to large weights
+
use early stopping during model training to stop training the model when test loss is becoming higher than the train loss
+
use torch.nn.Dropout() to randomly drop out some of the neurons to ensure the exisiting neurons will learn features without being too reliant on other neighbouring neurons in the network
+
I’ll try the early stopping or drop out method in future posts since current post is relatively long already…
+
+
To overcome underfitting:
+
+
increase training epochs
+
minimise regularisation
+
consider building a more complex or deeper neural network model
+
+
I’m trying to keep this post simple so have only used mean squared error (MSE) and mean absolute error (MAE) to evaluate the model which has made a prediction on the test set. The smaller the MSE, the less error the model has when making predictions. However this is not the only metric that will determine if a model is optimal for predictions, as I’ve also noticed that every time there’s a different train and test split, the MAE and MSE values will vary too, so it appears that some splits will generate smaller MSE and other splits will lead to larger MSE.
+
+
+Code
+
# torch.no_grad() - disable gradient calculations to reduce memory usage for inference (also like a decorator)
+with torch.no_grad():
+ predict_test = model(X_test.float())[:, 0]
+# Padding target tensor with set size of [(1, 2)] as input tensor size will vary
+# when notebook is re-run each time due to butina split with sample shuffling
+# so need to pad the target tensor accordingly
+ y_test_pad = F.pad(y_test, pad=(predict_test[None].shape[1] - y_test.shape[1], 0, 0, 0))
+ loss_new = loss_f(predict_test[None], y_test_pad)
+print(f"MSE for test set: {loss_new.item():.4f}")
+print(f"MAE for test set: {nn.L1Loss()(predict_test[None], y_test_pad).item():.4f}")
+
+
+
MSE for test set: 0.6576
+MAE for test set: 0.8070
-
Quinidine has 4 defined atom stereocentre count as per PubChem compound summary (as one of possible references for cross-checking) - this is based on the calculation for CHEMBL1294, which is the same as the quinidine spotted at index row 4. So I’m dropping the quinidine at index row 3 for now.
-
-
# Note: old index is unchanged for now (re-index later if needed)
-df_2d6_s_inh_p = df_2d6_s_inh_p.drop(labels =3)
-df_2d6_s_inh_p
I haven’t done feature standardisation for X_train which is to centre X_train mean and divide by its standard deviation, code may be like this, X_train_normalised = (X_train - np.mean(X_train))/np.std(X_train) (if used on training data, need to apply this to testing data too)
+
Training features are certainly too small, however, the main goal of this very first post is to get an overall idea of how to construct a baseline DNN regression model. There are lots of other things that can be done to the ADRs data e.g. adding more drug molecular features and properties. I have essentially only used the initial molecular fingerprints generated when doing the data split to add a bit of molecular aspect in the training dataset.
+
I haven’t taken into account the frequencies of words (e.g. same drug classes and same ADR terms across different drugs) in the training and testing data, however, the aim of this first piece of work is also not a semantic analysis in natural language processing so this might not be needed…
+
There may be other PyTorch functions that I do not yet know about that will deal with small datasets e.g. perhaps torch.sparse may be useful?… so this piece is certainly not the only way to do it, but one of the many ways to work with small data
+
+
+
+
+
Acknowledgements
+
I’m very thankful for the existence of these references, websites and reviewer below which have helped me understand (or scratch a small surface of) deep learning and also solve the coding issues mentioned in this post:
+
+
+
+
+ ]]>
+ Deep learning
+ Pytorch
+ RDKit
+ Pandas
+ Python
+ ChEMBL database
+ Toxicology
+ Metabolism
+ Cheminformatics
+ https://jhylin.github.io/Data_in_life_blog/posts/22_Simple_dnn_adrs/2_ADR_regressor.html
+ Tue, 07 Jan 2025 11:00:00 GMT
+
+
+ Notes on adverse drug reactions (ADRs) data
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/22_Simple_dnn_adrs/1_ADR_data.html
+ Here are the notes regarding the strong and moderate CYP3A4 substrates used in the data in the accompanying notebook.
+
Strength of evidence that the drug is metabolised by CYP3A4/5 (as quoted from above web link):
+
+
Strong Evidence: the enzyme is majorly responsible for drug metabolism.
+
Moderate Evidence: the enzyme plays a significant but not exclusive role in drug metabolism or the supporting literature is not extensive.
+
+
+
+
+
drug_class data sources
+
This information can be found in many national drug formularies, drug reference textbooks e.g. Martindale, American society of health-System pharmacists’ (ASHP) drug information (DI) monographs, PubChem, ChEMBL, FDA, Micromedex etc. or online drug resources such as Drugs.com. For the particular small dataset collected and used in the notebook, the following reference sources for ADRs also contain information on therapeutic drug classes.
using the health professional version for ADRs which usually contains ADR references from pharmaceutical manufacturers’ medicines information data sheets, ASHP DI monographs or journal paper references
+
+
2nd-line as separate data checks:
+
+
NZ formulary (nzf) - likely only available to NZ residents only; other national formularies should contain very similar drug information
drugs.com_uk_di - UK drug information section in Drugs.com (equivalent to pharmaceutical manufacturers’ medicines information data sheets)
+
+
two main types of occurrences/frequencies used:
+
^^ - common > 10%,
+
^ - less common 1% to 10%,
+
(not going to include other ones with lower incidences e.g. less common at 0.1% to 1%, rare for less than 0.1% etc.)
+
+
+
+
+
Exceptions or notes for ADRs
+
+
nausea and vomiting applies to many drugs so won’t be included (almost every drug will have these ADRs, they can be alleviated with electrolytes replacements and anti-nausea meds or other non-med options; rash on the other hand can sometimes be serious and life-threatening e.g. Stevens-Johnson syndrome)
+
similar or overlapping adverse effects will be removed to keep only one adverse effect for the same drug e.g. adverse skin reactions, rash, urticaria - rash and urticaria will be removed as allergic skin reactions encompass both symptoms
+
for ADR terms with similar meanings, e.g. pyrexia/fever - fever is used instead (only one will be used)
+
ADR mentioned in common ADR category and repeated in the less common one will have the ADR recorded in the higher incidence rate (at > 10%) only
+
some ADRs can be dose-related or formulations-related e.g. injection site irritations or allergic reactions caused by excipients/fillers (aim is to investigate the relationships between ADRs and drugs via computational tools e.g. any patterns between ADRs & drugs so dose/formulations-related ADRS will not be recorded here)
+
some postmarketing adverse effects are for different age populations e.g. paediatric patients of up to 12 years of age or elderly people - for now all of them are labelled as “(pm)” to denote postmarketing reports and are not differentiated in age groups
+
+
+
Notes for specific drugs
+
+
hydrocortisone (a moderate CYP3A4 substrate) has no reported ADR frequencies at all for its ADRs as they are entirely dependent on the dosage and duration of use (ADRs tend to be unnoticeable at appropriate low doses for short durations)
+
terfenadine (a strong CYP3A4 substrate) is actually withdrawn from the market in 1990s due to QT prolongations
+
lercanidipine (a moderate CYP3A4 substrate) has nil reported ADRs of more than 1% but has a few postmarketing reports recorded
+
telaprevir (a moderate CYP3A4 substrate) is usually administered within a combination therapy (e.g. along with peginterferon alfa and ribavirin)
+
quinine (a moderate CYP3A4 substrate) has all of its ADRs reported without frequencies. The most common ADRs are presented as a cluster of symptoms (known as cinchonism) and can occur during overdoses (usually very toxic) and also normal doses. These symptoms include “…tinnitus, hearing impairment, headache, nausea, vomiting, abdominal pain, diarrhoea, visual disturbances (including blindness), arrhythmias (which can have a very rapid onset), convulsions (which can be intractable), and rashes.” (as quoted from NZ formulary v150 - 01 Dec 2024)
+
ribociclib (a moderate CYP3A4 substrate) has a listed ADR of on-treatment deaths, which were found to be associated with patients also taking letrozole or fulvestrant at the same time and/or in patients with underlying malignancy
+
+
+
+
+
+
Abbreviations used
+
+
ws = withdrawal symptoms
+
ADH = antidiuretic hormone
+
pm = postmarketing reports
+
CNS = central nervous system
+
CFTR = cystic fibrosis transmembrane regulator
+
c_diff = Clostridioides/Clostridium difficile
+
ZE = Zollinger-Ellison
+
MTOR = mammalian target of rapamycin (protein kinase)
+
AST = aspartate transaminase/aminotransferase
+
ALT = alanine transaminase/aminotransferase
+
ALP = alkaline phosphatase
+
GGT = gamma-glutamyltransferase
+
RTI = respiratory tract infection
+
UTI = urinary tract infection
+
LDH = lactate dehydrogenase
+
dd = dose and duration-dependent
+
pm_HIV_pit = postmarketing reports for HIV protease inhibitor therapy
+
pm_hep_cyto = postmarketing reports in cancer patients where drug was taken with hepatotoxic/cytotoxic chemotherapy and antibiotics
+
+
+
+
+
+ ]]>
+ Data
+ Notes
+ https://jhylin.github.io/Data_in_life_blog/posts/22_Simple_dnn_adrs/1_ADR_data.html
+ Tue, 07 Jan 2025 11:00:00 GMT
+
+
+ Cytochrome P450 and small drug molecules
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/20_Cyp3a4_2d6_inh/1_CYP450_drugs.html
+
+
Outline
+
The main goal for this post is to have a preliminary look into two of the most common groups of cytochrome P450 (CYP) inhibitors with an initial aim to look for any maximum common substructures (MCS) in these compounds if present or applicable.
+
TL;DR - Many pharmaceuticals have different types of rings found in their chemical structures (Taylor, MacCoss, and Lawson 2014), so it is unsurprising that the MCSes found here are mainly rings. To further explore the underlying mechanisms of actions (e.g. putative or active binding sites) of these CYP inhibitors, the structures of CYPs should be examined at the same time ideally.
+
Below is a general outline of the post:
+
+
Small introduction on drug toxicology and metabolism regarding CYPs
+
+
citing data sources for drug structural alerts in ChEMBL
+
a thought on another drug metabolism data source
+
+
Data extraction using chembl_downloader
+
Data import and preprocessing
+
Quick check on structural validities
+
Further look into duplicated structures and stereochemistries
+
+
quinidine
+
itraconazole
+
+
Maximum common substructures
+
+
CYP3A4 inhibitors
+
CYP2D6 inhibitors
+
Some other interesting code re. MCS
+
+
Some small findings and possible future work
+
Acknowledgements
+
+
+
+
+
Some introductions
+
I initially want to work on something about drug toxicology without setting any goals or intentions on how this post will end (bit like a free-style post) and currently I feel it will be more to do with metabolism, which is also directly linked to drug toxicity, an important area not to ignore during any therapeutic drug discovery and developmental work.
+
This post is sort of inspired by a recent commentary that talks about “avoidome” and DMPK-related proteins to avoid (Fraser and Murcko 2024). I also happen to encounter two other blog posts (a post from D. Lowe, and another one from P. Kenny) that have provided reviews on this commentary recently with interesting view points.
+
In a very rough sense, three main areas have been looked at (not exhaustive) with the aim to create safer therapeutic drugs:
+
+
Structural alerts on compound substructures that are known to cause adverse drug effects or pan-assay interference compounds (PAINs)
+
Many have looked into structural alerts (an example repo: rd_filters). ChEMBL database has already had a cheminformatic utils web service developed that provides structural alert computations for compounds. There are most likely much more efforts than these ones.
+
Toxicophores in relation to human ether-a-go-go-related gene (hERG) potassium channel (related to structural alerts as well)
+
hERG potassium channel is also another frequently-looked-at aspect for drug toxicology due to its known effect leading to cardiac QT prolongations or more commonly known as arrhythmias (Curran et al. 1995).
+
CYP enzymes with the well-known ones as CYP3A4, 2D6, 1A2, 2C9 and 2C19
+
CYP450 enzymes play a key role in the metabolism and toxicology parts of the ADMET process of drugs. When a drug behaves like a cytochrome inhibitor, it inhibits the activity of a particular cytochrome enzyme e.g. CYP3A4 leading to a reduction of clearance of a particular therapeutic drug e.g. a CYP3A4 substrate such as apixaban, thus increasing its plasma concentration in vivo causing a higher chance of adverse effect (which in the context of apixaban, this means the poor person taking the apixaban may get excessive bleeding…).
+
+
Other useful categories involve drug-induced skin sensitisations and liver injuries and more.
+
My very inital naive thought is that if we can at least cover some of the drug toxicology part during drug design and discovery process, this may be able to save some resources along the way (obviously it won’t be this simple…). The main thing here is that it may still be useful and interesting to look into the relationship between CYP450 and small drug molecules - to see if there are anything worth further explorations. This post will start with the two largest groups of CYP inhibitors, so focussing on CYP3A4 and 2D6 first.
+
While my focus is only on a very small cohort of small molecules relating to only two CYPs, it is also worth noting that there are actually more CYPs present as well, for example, CYP1A1, 2A6, 2B6, 2C8, 2E1, 2J2, 3A5 (note: amlodipine is a moderate CYP3A5 inhibitor and will be looked at below), 3A7 and 4F2 (Guengerich 2020). The cited paper here also provides quite a comprehensive background on the history of CYP450 and their relevance to toxicities in drugs, so I won’t repeat them here.
+
+
+
More on structural alerts
+
I am only really curious about the data sources used to build these ChEMBL structural alerts, so below are some of my notes on these sources.
+
From ChEMBL 20, only 6 filters are present, as shown by this ChEMBL blogpost - it may appear that this blog post cites all 8 filters but in fact it only has 6. I’ve attempted to find out the sources of these ChEMBL structural alert sets, here they are:
From ChEMBL 20 to 23, there are 8 filters in total (agreed with rd_filters’ README.md that there aren’t many documentations about this in ChEMBL, as I’ve tried also), the sources of the two additional ones are as follow:
+
+
Inpharmatica - unable to find direct source initially but this is later confirmed as private communications between ChEMBL and Inpharmatica Ltd. in the earlier days - an older ChEMBL presentation on ChEMBL 09 mentions about this, and this is also further elaborated by this paper (Gaulton et al. 2016)
+
SureChEMBL (old link provided by the paper (Gaulton et al. 2016) also no longer exists)
+
+
RDKit (section on “Filtering unwanted substructures”) also has another NIH filter based on two other references (Jadhav et al. 2010) and (Doveston et al. 2015). At one point I’m so confused with this NIH filter here and the NIH MLSMR one above… they are actually different as different papers are cited.
+
RDKit also uses the above 8 filters mentioned in ChEMBL in its FilterCatalogs class currently. Brenk filter seems to be the same as the CHEMBL_Dundee one since both of them have quoted the same journal paper as reference. It’s also got a ZINC one I think. Before I get very carried away, I’ll stop searching for every structural alerts papers here as there are many in the literatures.
+
+
+
+
More on CYPs and ADMET
+
A bit of a sidetrack for this part (feel free to skip) as I come across a new paper online recently about using deep learning model for ADMET prediction which uses data from Therapeutics data commons (TDC). So while working on this relevant topic of CYP and ADMET (only the metabolism and toxicology parts), I just want to dig a bit deeper to see what sort of data are used by TDC.
+
The TDC ADME dataset, specifically the metabolism one on all five CYP isoenzymes (CYP2C19, 2D6, 3A4, 1A2 and 2C9), are all derived from a 2009 paper by Veith et al.. A closer look at this paper only seems to mention:
+
+
…we tested 17,143 samples at between seven and fifteen concentrations for all five CYP isozymes. The samples consisted of 8,019 compounds from the MLSMR including compounds chosen for diversity and rule-of-five compliance, 16 synthetic tractability, and availability; 6,144 compounds from a set of biofocused libraries which included 1,114 FDA-approved drugs; and 2,980 compounds from combinatorial libraries containing privileged structures targeted at GPCRs and kinases, and libraries of purified natural products or related structures…
+
+
If I go to its original journal paper site (the link provided was a NCBI one), there is only one additional Excel file with a long list of chemical scaffolds showing different CYP activities (no other supplementary information I can spot there). The only likely lists of compounds tested are shown in its figures 6 and 7 in the paper, where figure 7 is more relevant for drug-drug interactions. I then realise the proportions of FDA-approved drugs used and the rest of the molecules tested in this paper are also not very balanced (thinking along the line of approved drugs and non-approved drugs), and notice that what they are saying in its discussion about how they are not noticing the usual prominent activities of CYP3A4 and 2D6 in the compounds they’ve tested:
+
+
…It has been suggested that CYP 3A4 is the most prominent P450 isozyme in drug metabolism and hepatic distribution (Fig. 2b),25,26 but the drugs in our collection do not appear to have been optimized away from this activity. There has also been speculation that CYP 2D6 isozyme plays a prominent role in drug metabolism,27 but no difference in activity was observed between diversity compounds and approved drugs for this isozyme…
+
+
I wonder if this may be due to the imbalanced set of compounds used e.g. number of FDA-approved drug (smaller) vs. number of other compounds from other libraries (larger)…
+
I’ve also visited FDA’s website to look at how the CYP stories are compiled (FDA link). The in vitro inhibitors and clinical index inhibitors are not completely the same across all the CYPs. There are some overlappings in CYP3A4/5 and 2D6 for sure but definitely not exactly the same across all the documented CYPs in this FDA webpage.
+
So back to this new paper on predicting ADMET… how likely will it be useful in real-life hit/lead ADMET optimisation projects in drug discovery settings if the data source only involves a larger portion of non-approved drugs versus a smaller portion of actual FDA-approved drugs?… It just shows that there are a lot of things to think about in the DMPK/ADMET areas within drug discovery pipelines, as ultimately this is crucial to see if a candidate molecule will proceed or not (i.e. causing toxicity or not and whether it’s tolerable side effects or adverse or even life-threatening ones instead).
+
+
+
+
+
Extracting data
+
First step here is to import the following software packages in order to retrieve and work with ChEMBL data (again).
+
+
import pandas as pd
+import chembl_downloader
+from chembl_downloader import latest
+from rdkit import Chem
+from rdkit.Chem import Draw, AllChem
+# For maximum common substructures & labelling stereocentres
+from rdkit.Chem import rdFMCS, rdCIPLabeler
+from rdkit.Chem.Draw import IPythonConsole
+IPythonConsole.drawOptions.addAtomIndices =False
+# Change to false to remove stereochem labels
+IPythonConsole.drawOptions.addStereoAnnotation =True
+IPythonConsole.ipython_useSVG=True
+
+
+
# Latest version of ChEMBL
+latest_version = latest()
+print(f"The latest ChEMBL version is: {latest_version}")
+
+
The latest ChEMBL version is: 34
+
+
+
I’m using SQL via chembl_downloader to download approved drugs with their ChEMBL ID and equivalent canonical SMILES. All of the CYP3A4 and 2D6 inhibitors extracted from ChEMBL are based on the Flockhart table of drug interactions(Flockhart et al. 2021).
+
Note: Three other categories of medicines are not going to be looked at for now, which are the weak inhibitors, ones with in vitro evidence only and ones that are still pending reviews.
+
A bit about retrieving data here, the following may not be the best way to get the data, but I’ve somehow incorporated chembl_downloader into my own small piece of function code (see Python script named as “cyp_drugs.py” in the repo) to retrieve SMILES of approved drugs (other public databases may also work very well equally, but I’m used to using ChEMBL now as it’s easy to read and navigate).
+
Another possible way is to use get_target_sql() within chembl_downloader, e.g. using a specific CYP enzyme as the protein target to retrieve data, but it appears that there are no clear data marked to indicate the potency of CYP inhibition or induction (i.e. weak, moderate or strong) in the ChEMBL database (an example link for CYP2D6 in ChEMBL). The Flockhart table has clearly annotated each approved drug with journal paper citations so I decide to stick with the previous method.
+
+
## Main issue previously is with sql string - too many quotation marks!
+# e.g. WHERE molecule_dictionary.pref_name = '('KETOCONAZOLE', 'FLUCONAZOLE')'': near "KETOCONAZOLE": syntax error
+# Resolved issue by adding string methods e.g. strip() and replace() to sql query string
+
+from cyp_drugs import chembl_drugs
+
+# Get a list of strong cyp3a4 inhibitors
+# For the story on why I also added a weird spelling of "itraconzole", please see below.
+# and save as a tsv file
+df_3a4_strong_inh = chembl_drugs(
+"CERITINIB", "CLARITHROMYCIN", "DELAVIRIDINE", "IDELALISIB", "INDINAVIR", "ITRACONAZOLE", "ITRACONZOLE", "KETOCONAZOLE", "MIBEFRADIL", "NEFAZODONE", "NELFINAVIR", "RIBOCICLIB", "RITONAVIR", "SAQUINAVIR", "TELAPREVIR", "TELITHROMYCIN", "TUCATINIB", "VORICONAZOLE",
+#file_name="strong_3a4_inh"
+ )
+df_3a4_strong_inh.head()
Clearly, even if the SMILES of these two itraconzoles are not converted into RDKit molecules, we can probably tell one of them has stereochemistries and the other one is without due to the presence of “@” in the SMILES string for the one at index row 4. The output images show exactly that - one with chiral centres, where the other one doesn’t have any.
-
PubChem calculations have however generated different result for itraconazole. It seems it only has one defined atom stereocentre count and two undefined stereocentre counts (PubChem reference).
-
I’ve also noted that the two itraconzoles obtained from ChEMBL have different ChEMBL ID numbers (ChEMBL IDs: CHEMBL22587 and CHEMBL64391) to the one calculated in PubChem (ChEMBL ID: CHEMBL224725). So below I’ve looked into CHEMBL224725 first.
-
Then I realise that if I search for “itraconazole” directly in ChEMBL, only four entries will appear with ChEMBL IDs of CHEMBL64391, CHEMBL22587, CHEMBL882 and CHEMBL5090785, and there is no ChEMBL224725. This is all due to a small spelling error (which is most likely a typo by accident) of itraconazole - being spelled as “itraconzole”, which is also carried over into PubChem as well. I have checked again to make sure both “itraconzole” and the usual itraconazole are referring to the same chemical structure. Below are some screenshots showing the typo.
-
So to add this likely-mis-spelled “itraconzole” into the dataframe, I literally just add it into the SQL query above when obtaining drug information through chembl_downloader.
-
-
# SMILES of new addition - "itraconzole"
-df_3a4_s_inh_p.loc[6, "canonical_smiles"]
# Labelling stereocentres of new addition - "itraconzole"
-itracon_6 = Chem.MolFromSmiles("CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5COC(Cn6cncn6)(c6ccc(F)cc6F)O5)cc4)CC3)cc2)c1=O")
-rdCIPLabeler.AssignCIPLabels(itracon_6)
-itracon_6
-
-
-
-
-
-
There is only one stereocentre for “itraconzole”, which will match the CHEMBL224725 entry for “itraconzole” in PubChem. Without looking into other cross-referencing sources, and if only sticking with PubChem for now, I’ve then gone back to check all 3 (stereochemically-)different versions of itraconazole and found that the RDKit stereochemical calculations of these 3 itraconazoles have all basically matched their equivalent PubChem computations for atom sterecentre counts.
Dataframe df_3a4_s_inh_p (the preprocessed strong CYP3A4 inhibitors) containing 3 different itraconazoles is then updated below to remove two of the triplicated entries.
-
-
# Preprocessed df of strong cyp3a4 inhibitors
-df_3a4_s_inh_p.head(10)
-
-
-
-
-
-
-
-
-
chembl_id
-
pref_name
-
max_phase
-
canonical_smiles
-
rdkit_mol
-
standard_smiles
-
selfies
-
inchi
-
inchikey
-
-
-
-
-
0
-
CHEMBL2403108
-
CERITINIB
-
4.0
-
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
-
<rdkit.Chem.rdchem.Mol object at 0x135da19a0>
-
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
-
[C][C][=C][C][Branch2][Ring2][=Branch1][N][C][...
-
InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-...
-
VERWOWGGCGHDQE-UHFFFAOYSA-N
-
-
-
1
-
CHEMBL1741
-
CLARITHROMYCIN
-
4.0
-
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1b60>
-
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
-
[C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran...
-
InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21...
-
AGOYDEPGAOXOCK-KCBOHYOISA-N
-
-
-
2
-
CHEMBL2216870
-
IDELALISIB
-
4.0
-
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1a80>
-
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
-
[C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][...
-
InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-...
-
IFSDAJWBUCMOAH-HNNXBMFYSA-N
-
-
-
3
-
CHEMBL115
-
INDINAVIR
-
4.0
-
CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1ee0>
-
CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](...
-
[C][C][Branch1][C][C][Branch1][C][C][N][C][=Br...
-
InChI=1S/C36H47N5O4/c1-36(2,3)39-35(45)31-24-4...
-
CBVCZFGXHXORBI-PXQQMZJSSA-N
-
-
-
4
-
CHEMBL22587
-
ITRACONAZOLE
-
NaN
-
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5CO[C@](...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1cb0>
-
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5CO[C@](...
-
[C][C][C][Branch1][C][C][N][N][=C][N][Branch2]...
-
InChI=1S/C35H38Cl2N8O4/c1-3-25(2)45-34(46)44(2...
-
VHVPQPYKVGDNFY-ZPGVKDDISA-N
-
-
-
5
-
CHEMBL64391
-
ITRACONAZOLE
-
4.0
-
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1d90>
-
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn...
-
[C][C][C][Branch1][C][C][N][N][=C][N][Branch2]...
-
InChI=1S/C35H38Cl2N8O4/c1-3-25(2)45-34(46)44(2...
-
VHVPQPYKVGDNFY-UHFFFAOYSA-N
-
-
-
6
-
CHEMBL224725
-
ITRACONZOLE
-
NaN
-
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5COC(Cn6...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1f50>
-
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5COC(Cn6...
-
[C][C][C][Branch1][C][C][N][N][=C][N][Branch2]...
-
InChI=1S/C35H38F2N8O4/c1-3-25(2)45-34(46)44(24...
-
HUADITLKOCMHSB-RPOYNCMSSA-N
-
-
-
7
-
CHEMBL157101
-
KETOCONAZOLE
-
4.0
-
CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c...
-
<rdkit.Chem.rdchem.Mol object at 0x135da2030>
-
CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c...
-
[C][C][=Branch1][C][=O][N][C][C][N][Branch2][R...
-
InChI=1S/C26H28Cl2N4O4/c1-19(33)31-10-12-32(13...
-
XMAYWYJOQHXEEK-UHFFFAOYSA-N
-
-
-
8
-
CHEMBL45816
-
MIBEFRADIL
-
4.0
-
COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1e70>
-
COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c...
-
[C][O][C][C][=Branch1][C][=O][O][C@][Branch2][...
-
InChI=1S/C29H38FN3O3/c1-20(2)28-23-12-11-22(30...
-
HBNPJJILLOYFJU-VMPREFPWSA-N
-
-
-
9
-
CHEMBL623
-
NEFAZODONE
-
4.0
-
CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c...
-
<rdkit.Chem.rdchem.Mol object at 0x135da20a0>
-
CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c...
-
[C][C][C][=N][N][Branch2][Ring1][=Branch2][C][...
-
InChI=1S/C25H32ClN5O2/c1-2-24-27-31(25(32)30(2...
-
VRBKIVRKKCLPHA-UHFFFAOYSA-N
-
-
-
-
-
-
-
I’m keeping the one with max phase marked as 4.0 (due to the other two having “NaN” with no relevant medical or therapeutic indications data documented in PubChem).
-
-
# Note old index unchanged (re-index later if needed)
-# Dropping itraconazole at index rows 4 & 6
-df_3a4_s_inh_p = df_3a4_s_inh_p.drop(labels = [4, 6])
-df_3a4_s_inh_p.head(10)
-
-
-
-
-
-
-
-
-
chembl_id
-
pref_name
-
max_phase
-
canonical_smiles
-
rdkit_mol
-
standard_smiles
-
selfies
-
inchi
-
inchikey
-
-
-
-
-
0
-
CHEMBL2403108
-
CERITINIB
-
4.0
-
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
-
<rdkit.Chem.rdchem.Mol object at 0x135da19a0>
-
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
-
[C][C][=C][C][Branch2][Ring2][=Branch1][N][C][...
-
InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-...
-
VERWOWGGCGHDQE-UHFFFAOYSA-N
-
-
-
1
-
CHEMBL1741
-
CLARITHROMYCIN
-
4.0
-
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1b60>
-
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
-
[C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran...
-
InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21...
-
AGOYDEPGAOXOCK-KCBOHYOISA-N
-
-
-
2
-
CHEMBL2216870
-
IDELALISIB
-
4.0
-
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1a80>
-
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
-
[C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][...
-
InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-...
-
IFSDAJWBUCMOAH-HNNXBMFYSA-N
-
-
-
3
-
CHEMBL115
-
INDINAVIR
-
4.0
-
CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1ee0>
-
CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](...
-
[C][C][Branch1][C][C][Branch1][C][C][N][C][=Br...
-
InChI=1S/C36H47N5O4/c1-36(2,3)39-35(45)31-24-4...
-
CBVCZFGXHXORBI-PXQQMZJSSA-N
-
-
-
5
-
CHEMBL64391
-
ITRACONAZOLE
-
4.0
-
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1d90>
-
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn...
-
[C][C][C][Branch1][C][C][N][N][=C][N][Branch2]...
-
InChI=1S/C35H38Cl2N8O4/c1-3-25(2)45-34(46)44(2...
-
VHVPQPYKVGDNFY-UHFFFAOYSA-N
-
-
-
7
-
CHEMBL157101
-
KETOCONAZOLE
-
4.0
-
CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c...
-
<rdkit.Chem.rdchem.Mol object at 0x135da2030>
-
CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c...
-
[C][C][=Branch1][C][=O][N][C][C][N][Branch2][R...
-
InChI=1S/C26H28Cl2N4O4/c1-19(33)31-10-12-32(13...
-
XMAYWYJOQHXEEK-UHFFFAOYSA-N
-
-
-
8
-
CHEMBL45816
-
MIBEFRADIL
-
4.0
-
COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1e70>
-
COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c...
-
[C][O][C][C][=Branch1][C][=O][O][C@][Branch2][...
-
InChI=1S/C29H38FN3O3/c1-20(2)28-23-12-11-22(30...
-
HBNPJJILLOYFJU-VMPREFPWSA-N
-
-
-
9
-
CHEMBL623
-
NEFAZODONE
-
4.0
-
CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c...
-
<rdkit.Chem.rdchem.Mol object at 0x135da20a0>
-
CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c...
-
[C][C][C][=N][N][Branch2][Ring1][=Branch2][C][...
-
InChI=1S/C25H32ClN5O2/c1-2-24-27-31(25(32)30(2...
-
VRBKIVRKKCLPHA-UHFFFAOYSA-N
-
-
-
10
-
CHEMBL584
-
NELFINAVIR
-
4.0
-
Cc1c(O)cccc1C(=O)N[C@@H](CSc1ccccc1)[C@H](O)CN...
-
<rdkit.Chem.rdchem.Mol object at 0x135da2180>
-
Cc1c(O)cccc1C(=O)N[C@@H](CSc1ccccc1)[C@H](O)CN...
-
[C][C][=C][Branch1][C][O][C][=C][C][=C][Ring1]...
-
InChI=1S/C32H45N3O4S/c1-21-25(15-10-16-28(21)3...
-
QAGYKUNXZHXKMR-HKWSIXNMSA-N
-
-
-
11
-
CHEMBL3545110
-
RIBOCICLIB
-
4.0
-
CN(C)C(=O)c1cc2cnc(Nc3ccc(N4CCNCC4)cn3)nc2n1C1...
-
<rdkit.Chem.rdchem.Mol object at 0x135da22d0>
-
CN(C)C(=O)c1cc2cnc(Nc3ccc(N4CCNCC4)cn3)nc2n1C1...
-
[C][N][Branch1][C][C][C][=Branch1][C][=O][C][=...
-
InChI=1S/C23H30N8O/c1-29(2)22(32)19-13-16-14-2...
-
RHXHGRAEPCAFML-UHFFFAOYSA-N
-
-
-
-
-
-
-
After cleaning up the duplicated structures, below are the full sets of strong and moderate CYP3A4 inhibitors for structural checking.
MCS is something I’m interested in trying out so below are some examples of finding MCS in these CYP inhibitors. Please note that MCS may not be the most suitable strategy to look at these CYP inhibitors, I’m only using it to become a bit more familiar with it so that I can better understand MCS.
-
Some information regarding MCS in RDKit:
-
-
FindMCS is for 2 or more molecules and returns single-fragment MCS - based on FMCS algorithm (Dalke and Hastings 2013)
-
RascalMCES (maximum common edge substructures) is for 2 molecules only and returns multi-fragment MCES. A RDKit blog post by Dave Cosgrove talks about this in more details
I’m starting with the strong CYP3A4 inhibitors first.
-
-
# Get list of RDKit mols
-mols_s3a4 =list(df_3a4_s_inh_p["rdkit_mol"])
-
-# Find MCS in mols
-s3a4_mcs = rdFMCS.FindMCS(mols_s3a4)
-
-# Get images of highlighted MCS for strong CYP3A4 inhibitors
-Draw.MolsToGridImage(
- mols_s3a4,
- subImgSize=(400, 300),
- molsPerRow=2,
- legends =list(df_3a4_s_inh_p["pref_name"]),
- highlightAtomLists=[m.GetSubstructMatch(s3a4_mcs.queryMol) for m in mols_s3a4]
- )
-
-
-
-
-
You can get the number of atoms and bonds and also SMARTS string for the MCS like this below.
One way to customise MCS is via reducing molecule threshold to relax the MCS rule as suggested by the TeachOpenCADD reference above.
-
-
s3a4_mcs_80 = rdFMCS.FindMCS(mols_s3a4, threshold=0.8)
-
-Draw.MolsToGridImage(
- mols_s3a4,
- subImgSize=(400, 300),
- molsPerRow=2,
- legends =list(df_3a4_s_inh_p["pref_name"]),
- highlightAtomLists=[m.GetSubstructMatch(s3a4_mcs_80.queryMol) for m in mols_s3a4]
- )
-
-
-
-
-
-
# Without changing threshold
-s3a4_mcs1 = Chem.MolFromSmarts(s3a4_mcs.smartsString)
-
-# Lowered MCS threshold to 80% of mols
-s3a4_mcs2 = Chem.MolFromSmarts(s3a4_mcs_80.smartsString)
-
-Draw.MolsToGridImage([s3a4_mcs1, s3a4_mcs2], legends=["MCS1", "MCS2 with threshold = 0.8"])
-
-
-
-
-
Here the MCS differs between different MCS thresholds - when the threshold is not changed, it shows a partial contour of a ring structure, whereas the lowered threshold shows more of an alkyl chain structure.
-
-
-
-
Moderate CYP3A4 inhibitors
-
This is then followed by the moderate inhibitors for CYP3A4.
-
-
# Get list of RDKit mols
-mols_m3a4 =list(df_3a4_m_inh_p["rdkit_mol"])
-
-# Find MCS in mols
-m3a4_mcs = rdFMCS.FindMCS(mols_m3a4)
-
-# Get images of highlighted MCS for moderate CYP3A4 inhibitors
-Draw.MolsToGridImage(
- mols_m3a4,
- subImgSize=(400, 300),
- molsPerRow=2,
- legends =list(df_3a4_s_inh_p["pref_name"]),
- highlightAtomLists=[m.GetSubstructMatch(m3a4_mcs.queryMol) for m in mols_m3a4]
- )
-
-
-
-
-
Aromatic (and macrolide) rings are highlighted in the MCS for this group.
-
-
-
-
All CYP3A4 inhibitors
-
The moderate CYP3A4 inhibitors are then added next in order to see if MCS changes when looking at all of the CYP3A4 inhibitors.
-
-
# Concatenate dfs for moderate & strong CYP3A4 inhibitors
-df_3a4_all = pd.concat([df_3a4_s_inh_p, df_3a4_m_inh_p])
-# index un-changed for now
-print(df_3a4_all.shape)
-df_3a4_all.head(3)
-
-
(27, 9)
-
-
-
-
-
-
-
-
-
-
chembl_id
-
pref_name
-
max_phase
-
canonical_smiles
-
rdkit_mol
-
standard_smiles
-
selfies
-
inchi
-
inchikey
-
-
-
-
-
0
-
CHEMBL2403108
-
CERITINIB
-
4.0
-
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
-
<rdkit.Chem.rdchem.Mol object at 0x135da19a0>
-
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
-
[C][C][=C][C][Branch2][Ring2][=Branch1][N][C][...
-
InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-...
-
VERWOWGGCGHDQE-UHFFFAOYSA-N
-
-
-
1
-
CHEMBL1741
-
CLARITHROMYCIN
-
4.0
-
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1b60>
-
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
-
[C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran...
-
InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21...
-
AGOYDEPGAOXOCK-KCBOHYOISA-N
-
-
-
2
-
CHEMBL2216870
-
IDELALISIB
-
4.0
-
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
-
<rdkit.Chem.rdchem.Mol object at 0x135da1a80>
-
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
-
[C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][...
-
InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-...
-
IFSDAJWBUCMOAH-HNNXBMFYSA-N
-
-
-
-
-
-
-
-
mols_3a4_all =list(df_3a4_all["rdkit_mol"])
-
-# Find MCS for all CYP3A4 inhibitors
-all_3a4_mcs = rdFMCS.FindMCS(mols_3a4_all)
-
-# All CYP3A4 inhibitors
-Draw.MolsToGridImage(
- mols_3a4_all,
- subImgSize=(400, 300),
- molsPerRow=2,
- legends =list(df_3a4_all["pref_name"]),
- highlightAtomLists=[m.GetSubstructMatch(all_3a4_mcs.queryMol) for m in mols_3a4_all]
- )
-
-
-
-
-
It appears the MCS for most of them involves (partial) rings (e.g. cycloalkane, aromatic and macrolide ones). This result is basically not very different from when we’ve looked at the the CYP3A4 inhibitors separately in their moderate and strong potencies. The next thing I want to try is to add in the ring bonds matching.
-
-
# matching ring bonds
-all_3a4_mcs_ring = rdFMCS.FindMCS(mols_3a4_all, ringMatchesRingOnly=True)
-
-Draw.MolsToGridImage(
- mols_3a4_all,
- subImgSize=(400, 300),
- molsPerRow=2,
- legends =list(df_3a4_all["pref_name"]),
- highlightAtomLists=[m.GetSubstructMatch(all_3a4_mcs_ring.queryMol) for m in mols_3a4_all]
- )
-
-
-
-
-
The ring bond matching also shows a very similar result here as it only restricts the MCS matching to ring bonds only.
-
-
Some other interesting code re. MCS
-
One of the code examples I’d like to try next, in order to see if it makes any differences from above, is the code snippets shared by a RDKit post from Paolo Tosco - “New MCS features in 2023.09.1” - the “Custom Python AtomCompare and BondCompare classes” section.
-
Some notes from me:
-
-
AtomCompare & BondCompare - looking at elements/bond orders/aromaticities in ring systems or
-
custom subclasses in AtomCompare & BondCompare - looking at elements/bond orders/aromaticities in non-ring systems
-
-
i.e. customise parameters using rdFMCS.MCSParameters()
-
I’ll also attempt to add some code comments below to explain how the code works (anyone’s welcomed to report any issues or changes for this part).
-
-
Ring systems
-
Here, I’m trying the AtomCompare & BondCompare along with RingMatchesRingOnly and CompleteRingsOnly first.
-
-
-Code
-
## Customise MCS parameters
-# Initiate a MCSParameter object
-params = rdFMCS.MCSParameters()
-# Define atom typer to be used to compare elements within rdFMCS.AtomCompare class
-params.AtomTyper = rdFMCS.AtomCompare.CompareElements
-# Define bond typer to be used to compare bond orders within rdFMCS.BondCompare class
-params.BondTyper = rdFMCS.BondCompare.CompareOrder
-# RingMatchesRingOnly - ring atoms to match other ring atoms only
-# CompleteRingsOnly - match full rings only
-params.BondCompareParameters.RingMatchesRingOnly =True
-params.BondCompareParameters.CompleteRingsOnly =False
-
-all_3a4_ringMCS = rdFMCS.FindMCS(mols_3a4_all, params)
-
-Draw.MolsToGridImage(
- mols_3a4_all,
- subImgSize=(400, 300),
- molsPerRow=3,
- legends =list(df_3a4_all["pref_name"]),
- highlightAtomLists=[m.GetSubstructMatch(all_3a4_ringMCS.queryMol) for m in mols_3a4_all]
- )
-
-
-
-
-
-
Some MCS characteristics noted after trying this:
-
-
When both BondCompareParameters are set to true, no rings are highlighted (apart from ethyl chains being highlighted in every molecule)
-
When turning off CompleteRingsOnly, this allows partial rings to be shown in highlighted MCS
-
A similar output is generated when using ringMatchesRingOnly = True in FindMCS() earlier for all CYP3A4 inhibitors
-
-
-
Non-ring systems
-
The class code below is also borrowed from the RDKit blog post, which explains why it’s done as a custom “class” code and not a “function” one.
-
-
-Code
-
# I've had to think harder about the what the class code are doing below,
-# especially the differences between comparing bond orders and ring atoms...
-# I can only describe it as both a restrictive (matching non-ring bonds only)
-# and lenient (but still comparing ring atoms) process in order to cover the non-ring parts (?)
-
-# Compare bond orders outside ring systems using rdFMCS.MCSBondCompare
-# Using class code that will call a function object
-class CompareOrderOutsideRings(rdFMCS.MCSBondCompare):
-
-def__call__(self, p, mol1, bond1, mol2, bond2):
-
-# Get bonds 1 and 2 based on bond indices for mols 1 and 2
- b1 = mol1.GetBondWithIdx(bond1)
- b2 = mol2.GetBondWithIdx(bond2)
-# If bonds 1 and 2 are both in ring or if their bond types are the same
-if (b1.IsInRing() and b2.IsInRing()) or (b1.GetBondType() == b2.GetBondType()):
-# If using stereo matching parameter and not checking bond stereo descriptors,
-# return bonds that are not using stereo matching parameter
-if (p.MatchStereo andnotself.CheckBondStereo(p, mol1, bond1, mol2, bond2)):
-returnFalse
-# If using ring bonds-matching-ring bonds parameter
-# return mols/bonds that are part of a ring only
-if p.RingMatchesRingOnly:
-returnself.CheckBondRingMatch(p, mol1, bond1, mol2, bond2)
-returnTrue
-# Only match bonds that are not part of ring systems
-returnFalse
-
-# Compare atom elements outside ring systems using rdFMCS.MCSAtomCompare
-class CompareElementsOutsideRings(rdFMCS.MCSAtomCompare):
-
-def__call__(self, p, mol1, atom1, mol2, atom2):
-
-# Get atoms 1 and 2 based on atom indices for mols 1 and 2
- a1 = mol1.GetAtomWithIdx(atom1)
- a2 = mol2.GetAtomWithIdx(atom2)
-# If atomic numbers for both atoms are different and the atoms are not in ring systems,
-# return atoms that instead have the same atomic numbers and in rings systems
-if (a1.GetAtomicNum() != a2.GetAtomicNum()) andnot (a1.IsInRing() and a2.IsInRing()):
-returnFalse
-# If using matching atom chirality parameter and not checking both atoms have a chiral tag,
-# return atoms that are not using the matching atom chirality parameter
-if (p.MatchChiralTag andnotself.CheckAtomChirality(p, mol1, atom1, mol2, atom2)):
-returnFalse
-# If using ring bonds-matching-ring bonds parameter,
-# return mols/atoms that are part of a ring only
-if p.RingMatchesRingOnly:
-returnself.CheckAtomRingMatch(p, mol1, atom1, mol2, atom2)
-# Only match ring atoms against ring atoms
-returnTrue
-
-params_or = rdFMCS.MCSParameters()
-params_or.AtomTyper = CompareElementsOutsideRings()
-params_or.BondTyper = CompareOrderOutsideRings()
-params_or.BondCompareParameters.RingMatchesRingOnly =True
-params_or.BondCompareParameters.CompleteRingsOnly =True
-
-all_3a4_orMCS = rdFMCS.FindMCS(mols_3a4_all, params_or)
-
-Draw.MolsToGridImage(
- mols_3a4_all,
- subImgSize=(500, 400),
- molsPerRow=3,
- legends =list(df_3a4_all["pref_name"]),
- highlightAtomLists=[m.GetSubstructMatch(all_3a4_orMCS.queryMol) for m in mols_3a4_all]
- )
-
-
-
-
-
-
By using the suggested class code above, the MCS has indeed become larger, where a full ring is highlighted as the MCS.
-
A second code example from iwatobipen is to highlight molecular differences, which is different from highlighting only the MCSes. Alternative URL link and more examples can be accessed via RDKit Cookbook. Possible use cases of this code may be when dealing with a large set of analogues with changes in R-groups or during large compound screening and search (just as a few examples only). The main thing I can see from the code is that it begins with finding MCS for the input molecules, then uses SMARTS strings of the MCS in order to find atoms not within the MCS (using GetSubstructMatch() part) then highlights that part of the molecules.
-
-
-
-
All CYP2D6 inhibitors
-
Because of how the MCSes have turned out for CYP3A4 inhibitors above, I think I should just look at CYP2D6 inhibitors as a whole here. First thing is to combine the dataframes between the moderate and strong inhibitor groups.
-
-
# Concatenate dfs
-df_2d6_all = pd.concat([df_2d6_s_inh_p, df_2d6_m_inh_p])
-# index un-changed for now
-print(df_2d6_all.shape)
-df_2d6_all.head()
-
-
(14, 9)
-
-
-
-
-
-
-
-
-
-
chembl_id
-
pref_name
-
max_phase
-
canonical_smiles
-
rdkit_mol
-
standard_smiles
-
selfies
-
inchi
-
inchikey
-
-
-
-
-
0
-
CHEMBL894
-
BUPROPION
-
4.0
-
CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1
-
<rdkit.Chem.rdchem.Mol object at 0x135da3530>
-
CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1
-
[C][C][Branch1][#Branch2][N][C][Branch1][C][C]...
-
InChI=1S/C13H18ClNO/c1-9(15-13(2,3)4)12(16)10-...
-
SNPPWIUOZRMYNY-UHFFFAOYSA-N
-
-
-
1
-
CHEMBL41
-
FLUOXETINE
-
4.0
-
CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1
-
<rdkit.Chem.rdchem.Mol object at 0x135da3290>
-
CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1
-
[C][N][C][C][C][Branch2][Ring1][Ring2][O][C][=...
-
InChI=1S/C17H18F3NO/c1-21-12-11-16(13-5-3-2-4-...
-
RTHCYVBBDHJXIQ-UHFFFAOYSA-N
-
-
-
2
-
CHEMBL490
-
PAROXETINE
-
4.0
-
Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1
-
<rdkit.Chem.rdchem.Mol object at 0x135da3bc0>
-
Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1
-
[F][C][=C][C][=C][Branch2][Ring1][#Branch2][C@...
-
InChI=1S/C19H20FNO3/c20-15-3-1-13(2-4-15)17-7-...
-
AHOUBRCZNHFOSL-YOEHRIQHSA-N
-
-
-
4
-
CHEMBL1294
-
QUINIDINE
-
4.0
-
C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc...
-
<rdkit.Chem.rdchem.Mol object at 0x135da2d50>
-
C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc...
-
[C][=C][C@H1][C][N][C][C][C@H1][Ring1][=Branch...
-
InChI=1S/C20H24N2O2/c1-3-13-12-22-9-7-14(13)10...
-
LOUPRKONTZGTKE-LHHVKLHASA-N
-
-
-
0
-
CHEMBL254328
-
ABIRATERONE
-
4.0
-
C[C@]12CC[C@H](O)CC1=CC[C@@H]1[C@@H]2CC[C@]2(C...
-
<rdkit.Chem.rdchem.Mol object at 0x135dd0270>
-
C[C@]12CC[C@H](O)CC1=CC[C@@H]1[C@@H]2CC[C@]2(C...
-
[C][C@][C][C][C@H1][Branch1][C][O][C][C][Ring1...
-
InChI=1S/C24H31NO/c1-23-11-9-18(26)14-17(23)5-...
-
GZOSMCIZMLWJML-VJLLXTKPSA-N
-
-
-
-
-
-
-
-
mols_all_2d6 =list(df_2d6_all["rdkit_mol"])
-
-# Find MCS for all CYP2D6 inhibitors
-all_2d6_mcs = rdFMCS.FindMCS(mols_all_2d6)
-
-# Get images of highlighted MCS for all CYP2D6 inhibitors
-Draw.MolsToGridImage(
- mols_all_2d6,
- subImgSize=(400, 300),
- molsPerRow=2,
- legends =list(df_2d6_all["pref_name"]),
- highlightAtomLists=[m.GetSubstructMatch(all_2d6_mcs.queryMol) for m in mols_all_2d6]
- )
-
-
-
-
-
Again, only a phenyl ring is highlighted as the MCS so this is not informative at all.
-
-
-
-
-
Some findings and future work
-
Below are some of my small findings and thoughts.
-
-
As mentioned at the very top in the outline section, rings (heterocycles, aromatic or fused ones) are everywhere in the MCSes for CYP3A4 and CYP2D6 inhibitors, and they are very common in known approved drugs
-
Looking at CYP structures in relation to these inhibitors may be more meaningful and also may reveal more insights about possible binding sites or mechanisms of actions of CYP inhibitions for these compounds. This may also be further explored in parallel to the actual drug targets of these CYP inhibitors e.g. binding site on CYP isoenzyme versus binding site on drug target protein as there are different classes of drugs within these CYP inhibitors. For example, some of CYP2D6 inhibitors are drugs acting on central nervous system (about 9 out of 14 drugs) - e.g. bupropion, fluoxetine, paroxetine, clobazam, doxepin, duloxetine, locaserin, moclobemide and rolapitant. For the CYP3A4 inhibitors, they are in several different therapeutic classes e.g. antivirals, antifungals, antibacterials, kinase inhibitors etc.
-
It may be a bit more interesting to compare the MCSes between CYP3A4 and CYP2D6 substrates (adding in other substrates for other CYPs)
-
Future posts may involve looking at CYP substrates using different cheminformatics strategies or doing molecular docking within a notebook setting (has this been done before?)
-
CYP inducers are a different story as they tend to increase drug metabolisms via CYP inductions, which are more likely to do with losing therapeutic effects than gaining adverse effects, so they may be looked at further down the line
-
MCS may not be absolutely useful in every scenario as I think it aims to look for the most maximum common substructures within a set of molecules, so not every molecule will be able to have a MCS shown (e.g. in a very diverse chemical set), other similarity searching techniques should probably be used instead if needed
-
-
-
-
-
Acknowledgements
-
Thanks to every contributor, developer or authors of every software package used in this post, and also the online communities behind them. Before I forget, the thanks would also be extended to the authors of the journal papers cited in this post. Lastly, special thanks to Noel O’Boyle for being very patient with reading my awfully-long old draft earlier and pointed out some useful things to note and change (I kind of got lost when writing the draft… due to it being a “free-style post”, I should avoid doing this again).
-
-
-
-
-
-
References
-
-Curran, Mark E, Igor Splawski, Katherine W Timothy, G.Michael Vincen, Eric D Green, and Mark T Keating. 1995. “A Molecular Basis for Cardiac Arrhythmia: HERG Mutations Cause Long QT Syndrome.”Cell 80 (5): 795–803. https://doi.org/10.1016/0092-8674(95)90358-5.
-
-
-Dalke, Andrew, and Janna Hastings. 2013. “FMCS: A Novel Algorithm for the Multiple MCS Problem.”Journal of Cheminformatics 5 (S1). https://doi.org/10.1186/1758-2946-5-s1-o6.
-
-
-Doveston, Richard G., Paolo Tosatti, Mark Dow, Daniel J. Foley, Ho Yin Li, Amanda J. Campbell, David House, Ian Churcher, Stephen P. Marsden, and Adam Nelson. 2015. “A Unified Lead-Oriented Synthesis of over Fifty Molecular Scaffolds.”Organic & Biomolecular Chemistry 13 (3): 859–65. https://doi.org/10.1039/c4ob02287d.
-
-Gaulton, Anna, Anne Hersey, Michał Nowotka, A. Patrícia Bento, Jon Chambers, David Mendez, Prudence Mutowo, et al. 2016. “The ChEMBL database in 2017.”Nucleic Acids Research 45 (D1): D945–54. https://doi.org/10.1093/nar/gkw1074.
-
-
-Guengerich, F. Peter. 2020. “A History of the Roles of Cytochrome P450 Enzymes in the Toxicity of Drugs.”Toxicological Research 37 (1): 1–23. https://doi.org/10.1007/s43188-020-00056-z.
-
-
-Jadhav, Ajit, Rafaela S. Ferreira, Carleen Klumpp, Bryan T. Mott, Christopher P. Austin, James Inglese, Craig J. Thomas, David J. Maloney, Brian K. Shoichet, and Anton Simeonov. 2010. “Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Artifacts in a Screen for Inhibitors of a Thiol Protease.”Journal of Medicinal Chemistry 53 (1): 37–51. https://doi.org/10.1021/jm901070c.
-
-
-Taylor, Richard D., Malcolm MacCoss, and Alastair D. G. Lawson. 2014. “Rings in Drugs.”Journal of Medicinal Chemistry 57 (14): 5845–59. https://doi.org/10.1021/jm4017625.
-
-
]]>
- Metabolism
- Toxicology
- Structural alerts
- Pandas
- RDKit
- ChEMBL database
- Python
- https://jhylin.github.io/Data_in_life_blog/posts/20_Cyp3a4_2d6_inh/1_CYP450_drugs.html
- Wed, 21 Aug 2024 12:00:00 GMT
-
-
-
- Boosted trees
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/19_ML2-3_Boosted_trees/1_adaboost_xgb.html
-
-
Some introductions
-
I’ve somehow promised myself to do a tree series on machine learning and glad I’ve made it to the boosted trees part (it took a while…). This is also likely my last post on this topic for now as there are other things I want to explore in the near future. Hopefully this is somewhat useful for anyone who’s new to this.
-
-
-
AdaBoost
-
Adaptive Boost or AdaBoost has originated from Robert E. Schapire in 1990 (Schapire 1990), (Raschka, Liu, and Mirjalili 2022), and was further introduced in 1996 by Robert Schapire and Yoav Freund at a conference which also led to a publication (Freund and Schapire 1997).
-
As quoted from scikit-learn, an AdaBoost algorithm is doing this:
-
-
…fit a sequence of weak learners (i.e., models that are only slightly better than random guessing, such as small decision trees) on repeatedly modified versions of the data.
-
-
-
-
-
-
-
-Note
-
-
-
-
Weak learner means an ensemble of very simple base classifiers such as decision tree stumps (Raschka, Liu, and Mirjalili 2022)
-
-
-
During the process of running the algorithm, increased weights are given to the incorrectly predicted samples at each iteration, and less weights are given to the correctly predicted ones. This then forces the AdaBoost models to focus more on the less accurately predicted samples with the aim to improve ensemble performance. The predictions from these iterations are combined to produce a final prediction via a weighted majority vote style, which is a well-known signature of tree models. Overall, AdaBoost algorithm can be used for classification or regression problems. The main difference between bagging and boosting is that boosting only uses random subsets of training samples drawn from the training dataset without any replacements (Raschka, Liu, and Mirjalili 2022). One caveat to note is that AdaBoost tend to overfit training data (high variance).
-
Parameters to tune:
-
-
n_estimators - number of weak learners
-
learning_rate - contributions of weak learners in the final combination
-
max_depth - depth of trees
-
min_samples_split - minimum required number of samples to consider a split
-
-
-
-
-
Gradient boosted trees
-
Essentially a similar concept is behind gradient boosted trees where a series of weak learners is trained in order to create a stronger ensemble of models (Raschka, Liu, and Mirjalili 2022). However, some differences between these two types of boosted trees (e.g. AdaBoost and XGBoost) should be noted, and rather than describing them in a paragraph, I’ve summarised them in a table below.
-
-
-
-
-
-
-
AdaBoost
-
XGBoost
-
-
-
-
-
trains weak learners based on errors from previous decision tree stump
-
trains weak learners that are deeper than decision tree stumps with a max depth of 3 to 6 (or max number of leaf nodes from 8 to 64)
-
-
-
uses prediction errors to calculate sample weights and classifier weights
-
uses prediction errors directly to produce the target variable to fit the next tree
-
-
-
uses individual weighting terms for each tree
-
uses a global learning rate for each tree
-
-
-
-
Differences between XGBoost and AdaBoost (Raschka, Liu, and Mirjalili 2022)
-
-
XGBoost or extreme gradient boosting (Chen and Guestrin 2016) is one of the most commonly used open-source packages, originally developed at the University of Washington by T. Chen and C. Guestrin, that uses stochastic gradient boosting to build an ensemble of predictive models.
Main parameters to tune as suggested by (Bruce, Bruce, and Gedeck 2020):
-
-
subsample - controls fraction of observations that should be sampled at each iteration or a subsample ratio of the training instance (as per XGBoost’s scikit-learn API). This is similar to how a random forest operates but without the sample replacement part
-
eta (in XGBoost) or learning_rate (in scikit-learn wrapper interface for XGBoost) - a shrinkage factor applied to alpha (a factor derived from weighted errors) in the boosting algorithm or it simply may be more easily understood as the boosting learning rate used to prevent overfitting
-
-
There are of course a whole bunch of other XGBoost parameters that can be tuned, and in order to keep this post at a reasonable reading length, I won’t go through every single one of them, but see this link as an example parameter set for XGBClassifier().
-
In scikit-learn, there are also two types of gradient boosted tree methods, GradientBoostingClassifer() and HistGradientBoostingClassifier(), in its sklearn.ensemble module (note: equivalent regressor class also available). One way to choose between them is to check sample size first. GradientBoostingClassifer() class is likely better when there is only a small sample size (e.g. when number of sample is less than 10,000), while HistGradientBoostingClassifier() class is likely better when your sample size is at least 10,000+ or more. The HistGradientBoostingClassifier() is a histogram-based gradient boosting classification tree that is mainly inpired by LightGBM.
-
-
-
-
-
A demo
-
In the example below, I’m only using AdaBoost classifier and XGBoost classifier for now. Please note that the dataset used here is very small and the example is likely not going to reflect real-life use case completely (use with care).
xgboost version used: 2.0.3
-scikit-learn version used: 1.5.0
-rdkit version used: 2023.09.6
-
-
-
-
# Show Python version used
-print(sys.version)
-
-
3.11.0 (v3.11.0:deaf509e8f, Oct 24 2022, 14:43:23) [Clang 13.0.0 (clang-1300.0.29.30)]
-
-
-
-
-
-
Data source
-
Data source is based on ChEMBL database version 33 (as shown by the file name below, “chembl_d_ache_33”), which was downloaded previously from last post (on random forest classifier) by using chembl_downloader.
-
-
from pathlib import Path
-
-# Pick any directory, but make sure it's relative to your home directory
-directory = Path.home().joinpath(".data", "blog")
-# Create the directory if it doesn't exist
-directory.mkdir(exist_ok=True, parents=True)
-
-# Create a file path that corresponds to the previously cached ChEMBL data
-path = directory.joinpath(f"chembl_d_ache_33.tsv")
-
-# alternative way to download latest ChEMBL version
-# please see post link - https://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/2_random_forest_classifier.html#data-retrieval-using-chembl_downloader for details
-# note: need to specify late_version = latest() first
-# path = directory.joinpath(f"chembl_d_ache_{latest_version}.tsv")
-
-if path.is_file():
-# If the file already exists, load it
- df_ache = pd.read_csv(path, sep=',')
-else:
-# If the file doesn't already exist, make the query then cache it
- df_ache = chembl_downloader.query(sql)
- df_ache.to_csv(path, sep=",", index=False)
The definition of “NaN” assigned to max_phase indicates that compounds labelled as “NaN” or “null” have no evidence of showing they’ve reached clinical trials yet, but I’m still keeping them in the dataset (this can also be excluded depending on project goals).
-
A max_phase of -1 is assigned to molecules with unknown clinical phase status (ChEMBL reference), for which I’ll drop for this particular experiment.
-
-
# Fill "NaNs" as "0" first
-df_ache.fillna({"max_phase": 0}, inplace=True)
-
-
-
# Select only mols with max_phase of 0 and above
-df_ache = df_ache[(df_ache["max_phase"] >=0)]
# Convert max_phase from float to int for the ease of reading predicted outcomes,
-# otherwise it'll look like "4., 2., 4., ..."
-df_ache = df_ache.astype({"max_phase": int, "canonical_smiles": "string"})
Please note: the only molecule with max_phase of “0.5” was converted into “0” after I’ve converted the datatype of max_phase from float to integer (I’ve left it deliberately like this since this is only a demonstration on using scikit_learn’s pipeline along with scikit_mol, but in reality this should be handled with care i.e. don’t discard it as different max phase values have different meanings!). Therefore the following max_phase value count will have 6411 molecules with max_phase “0”, rather than the previous number of 6410.
Binary classification has been used in my previous posts, e.g. target as max_phase 4 (re-labelled as “1”) with training set of max_phase “null” re-labelled as “0” with their different RDKit molecular features. This time I’ll be using multi-class classification to predict a training set of molecules containing max_phase of 0, 1, 2, 3 and 4.
-
-
-
Define X and y variables
-
-
# A sanity check and view on the original dataset
-df_ache
-
-
-
-
-
-
-
-
-
assay_chembl_id
-
target_type
-
tax_id
-
chembl_id
-
canonical_smiles
-
molecule_chembl_id
-
max_phase
-
standard_type
-
pchembl_value
-
-
-
-
-
0
-
CHEMBL1909212
-
SINGLE PROTEIN
-
9606
-
CHEMBL220
-
CC/C(=C(/CC)c1ccc(O)cc1)c1ccc(O)cc1
-
CHEMBL411
-
4
-
IC50
-
4.59
-
-
-
1
-
CHEMBL1003053
-
SINGLE PROTEIN
-
9606
-
CHEMBL220
-
COc1c2occc2cc2ccc(=O)oc12
-
CHEMBL416
-
4
-
IC50
-
4.27
-
-
-
2
-
CHEMBL2406149
-
SINGLE PROTEIN
-
9606
-
CHEMBL220
-
COc1c2occc2cc2ccc(=O)oc12
-
CHEMBL416
-
4
-
IC50
-
6.12
-
-
-
4
-
CHEMBL3071788
-
SINGLE PROTEIN
-
9606
-
CHEMBL220
-
O=c1cc(-c2ccc(O)cc2)oc2cc(O)cc(O)c12
-
CHEMBL28
-
0
-
IC50
-
7.92
-
-
-
5
-
CHEMBL1119333
-
SINGLE PROTEIN
-
9606
-
CHEMBL220
-
CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1
-
CHEMBL41
-
4
-
IC50
-
6.89
-
-
-
...
-
...
-
...
-
...
-
...
-
...
-
...
-
...
-
...
-
...
-
-
-
7139
-
CHEMBL5216374
-
SINGLE PROTEIN
-
9606
-
CHEMBL220
-
CCC(C)(C)NCC(O)c1cc(O)cc(OC(=O)N(C)C)c1
-
CHEMBL5220560
-
0
-
IC50
-
4.70
-
-
-
7140
-
CHEMBL5216425
-
SINGLE PROTEIN
-
9606
-
CHEMBL220
-
CC(C)(C)OC(=O)Nc1ccc(O)c(C(=O)NCCCN2CCCCC2)c1
-
CHEMBL5220695
-
0
-
Ki
-
6.92
-
-
-
7141
-
CHEMBL5216408
-
SINGLE PROTEIN
-
9606
-
CHEMBL220
-
O=C1CCOc2cc(OCCCCCSC(=S)N3CCCCC3)ccc21
-
CHEMBL5220742
-
0
-
IC50
-
7.00
-
-
-
7142
-
CHEMBL5218078
-
SINGLE PROTEIN
-
9606
-
CHEMBL220
-
Cn1ccc2cc(-c3cnc4ccc(C(=O)N5CCCCC5)cc4n3)ccc2c1=O
-
CHEMBL5220884
-
0
-
IC50
-
5.27
-
-
-
7143
-
CHEMBL5216374
-
SINGLE PROTEIN
-
9606
-
CHEMBL220
-
COC(=O)c1cc2c(OC(=O)N(C)C)cccc2[n+]([11CH3])c1
-
CHEMBL5220983
-
0
-
IC50
-
7.70
-
-
-
-
7075 rows × 9 columns
-
-
-
-
-
X = df_ache.canonical_smiles
-y = df_ache.max_phase
This post is going to focus on Scikit_mol which has a manual way to handle SMILES errors, as shown in the code below. Another useful way to deal with SMILES errors is Molpipeline’s SMILES error handling, with an example shown in one of its notebooks. The main difference from what I can see, even though I haven’t got to use it yet, is that Molpipeline takes into account of all the invalid SMILES by giving each invalid SMILES a “NaN” label in the pipeline process - this maintains the matrix shape and good for tracking down the problematic SMILES (molecules).
Also, manually checking for any “NaNs” in the canonical SMILES column (X variable), since AdaBoost classifier won’t accept missing values in the dataset, but if using HistGradientBoostingClassifier() instead, it should take care of the native NaNs.
-
-
print(f"{df_ache.canonical_smiles.isna().sum()} out of {len(df_ache)} SMILES failed in conversion")
-
-
0 out of 7075 SMILES failed in conversion
-
-
-
There are other ways to deal with NaNs with a few examples provided by scikit-learn. However, with regards to drug discovery data, there are probably more caveats that need to be taken during data preprocessing (I’m also still exploring this too).
-
-
-
-
Split data
-
Randomly splitting data this time.
-
-
# Found a silly error when naming X, y train/test sets!
-# Remember to name them in this order: X_train, X_test, y_train, y_test
-# otherwise model fitting won't work...
-X_train, X_test, y_train, y_test = train_test_split(X_valid, y, test_size=0.2, random_state=3)
-
-
-
-
-
-
Create pipelines
-
The aim is to create pipeline(s) using scikit-learn.
-
-
-
AdaBoost classifier
-
The original plan is to chain an AdaBoost classifier, XGBoost classifier, along with Scikit-mol transformers all at once. However, it turns out that I’m building two separate pipelines of AdaBoost classifier and XGBoost classifier so that I can compare the difference(s) between them, and this also serves better for the purpose of this post really.
-
This is also the time to think about generating molecular features for model training. Choosing data features such as fingerprints (e.g. Morgan fingerprints which is usually best for larger dataset) or RDKit 2D descriptors (which is useful for smaller datasets) or others. For RDKit 2D descriptors, Scikit_mol has integrated RDKit’s rdkit.Chem.Descriptors module and rdkit.ML.Descriptors.MoleculeDescriptors module within its MolecularDescriptorTransformer().
-
Some useful links regarding building pipelines in scikit-learn and also another reference notebook on when to use parallel calculations for different molecular features:
For the first sample pipeline I’m building below, I’ve noticed that not all of the 209 RDKit 2D descriptors can be used for AdaBoost classifier, as some of the descriptors will have values of “0” and AdaBoost classifier will not be able to take care of them. Therefore, I’m only using a small selection of descriptors only, but HistGradientBoostingClassifier() should be able to take into account of NaNs and can be chained to include all descriptors in a pipeline.
-
The following is an example of building a scikit_learn pipeline by using AdaBoost classifier model, along with Scikit_mol’s transformers for multi-class max_phase predictions with training set consisting of molecules with max_phase 0, 1, 2, 3 and 4. I’ve used Morgan fingerprints instead eventually so that’ll be shown in the following pipeline code, but I’ve also kept the RDKit 2D descriptor option on (just need to uncomment to run).
-
-
# Set parameters for RDKit 2D descriptors
-# params_rdkit2d = {
-# "desc_list": ['HeavyAtomCount', 'FractionCSP3', 'RingCount', 'MolLogP', 'MolWt']
-# }
-
-# Set parameters for adaboost model
-params_adaboost = {
-"estimator": DecisionTreeClassifier(max_depth =3),
-# default: n_estimators = 50, learning_rate = 1.0 (trade-off between them)
-"n_estimators": 80,
-"learning_rate": 0.2,
-# SAMME (Stagewise Additive Modeling using a Multi-class Exponential loss function) algorithm
-# for multi-class classification
-"algorithm": "SAMME",
-"random_state": 2,
- }
-
-# Building AdaBoostClassifier pipeline
-mlpipe_adaboost = make_pipeline(
-# Convert SMILES to RDKit molecules
- SmilesToMolTransformer(),
-# Molecule standardisations
- Standardizer(),
-## A choice of using either Morgan fingerprints or RDKit 2D descriptors:
-# Generate MorganFingerprintTransformer()
- MorganFingerprintTransformer(useFeatures=True),
-# Generate RDKit2D descriptors
-#MolecularDescriptorTransformer(**params_rdkit2d),
-# Scale variances in descriptor data
- StandardScaler(),
-# Apply adaptive boost classifier
- AdaBoostClassifier(**params_adaboost)
-)
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
An interactive pipeline diagram should show with a status of “not fitted” if you hover the mouse over the “i” logo on top right. The pipeline is then fitted onto the training sets.
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
The pipeline status should now show a “fitted” message if hovering over the same “i” logo. Then the pipeline is used on the X_test (testing X set) to predict the target (max_phase) variable.
-
-
mlpipe_adaboost.predict(X_test)
-
-
array([0, 0, 0, ..., 0, 0, 0])
-
-
-
-
-
-
XGBoost classfier
-
The following code snippet is an example of a scikit_learn pipeline using Scikit_mol’s transformers and XGBoost classifier. One nice thing about XGBoost is that is has scikit_learn interface so that it can be integrated into the scikit-learn pipeline and Scikit_mol’s transformers, which is what I’ve tried below.
-
-
# Set parameters for xgboost model
-params_xgboost = {
-"n_estimators": 100,
-"max_depth": 3,
-# For multi-class classification, use softprob for loss function (learning task parameters)
-# source: https://xgboost.readthedocs.io/en/latest/parameter.html#learning-task-parameters
-"objective": 'multi:softprob',
-"learning_rate": 0.1,
-"subsample": 0.5,
-"random_state": 2
- }
-
-# Building XGBoostClassifier pipeline
-mlpipe_xgb = make_pipeline(
-# Convert SMILES to RDKit molecules
- SmilesToMolTransformer(),
-# Molecule standardisations
- Standardizer(),
-## A choice of using either Morgan fingerprints or RDKit 2D descriptors:
-# Generate MorganFingerprintTransformer()
- MorganFingerprintTransformer(useFeatures=True),
-# Generate RDKit2D descriptors
-#MolecularDescriptorTransformer(**params_rdkit2d),
-# Scale variances in descriptor data
- StandardScaler(),
-# XGBoost classifier
- XGBClassifier(**params_xgboost)
-)
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
One can never just leave the process of building a machine learning model without evaluating it. Although what I have done below is probably very minimal but it’s somewhat a starting point to think about how good the model is.
-
-
from sklearn.metrics import accuracy_score
-
-# Following misclassification score function code borrowed and adapted from:
-# https://scikit-learn.org/stable/auto_examples/ensemble/plot_adaboost_multiclass.html#sphx-glr-auto-examples-ensemble-plot-adaboost-multiclass-py
-
-def misclassification_error(y_true, y_pred):
-return1- accuracy_score(y_true, y_pred)
-
-mlpipe_adaboost_misclassification_error = misclassification_error(
- y_test, mlpipe_adaboost.fit(X_train, y_train).predict(X_test)
-)
-
-mlpipe_xgb_misclassifiaction_error = misclassification_error(
- y_test, mlpipe_xgb.fit(X_train, y_train).predict(X_test)
-)
-
-print("Training score for mlpipe_adaboost: "f"{mlpipe_adaboost.score(X_train, y_train):0.2f}")
-print("Testing score for mlpipe_adaboost: "f"{mlpipe_adaboost.score(X_test, y_test):0.2f}")
-print("AdaBoostClassifier's misclassification_error: "f"{mlpipe_adaboost_misclassification_error:0.3f}")
-
-print("Training score for mlpipe_xgb: "f"{mlpipe_xgb.score(X_train, y_train):0.2f}")
-print("Testing score for mlpipe_xgb: "f"{mlpipe_xgb.score(X_test, y_test):0.2f}")
-print("XGBClassifier's missclassification_error: "f"{mlpipe_xgb_misclassifiaction_error:0.3f}")
-
-
Training score for mlpipe_adaboost: 0.97
-
-
-
Testing score for mlpipe_adaboost: 0.97
-AdaBoostClassifier's misclassification_error: 0.028
-
-
-
Training score for mlpipe_xgb: 0.99
-
-
-
Testing score for mlpipe_xgb: 0.99
-XGBClassifier's missclassification_error: 0.014
-
-
-
It appears that XGBoost model obtained a better prediction accuracy than the AdaBoost one (although the models are built in a very simple way, but it still shows the slight difference in performance). The training data being used here is also very imbalanced with a lot of them being max_phase of “0” than “4”, and with max_phase “4” being our ultimate aim, the dataset used above is really for demonstration only. Also, since this post is already quite long, I’d rather not make this post into a gigantic tl;dr, so for the imbalanced data discussion and exploration, my previous posts have tried to touch on this topic - “Random forest” and “Random forest classifier”.
-
-
-
-
Hyperparameter tuning for XGBoost classifier
-
For XGBoost, one of the main things is to minimise model overfitting where several parameters will play important roles to achieve this. For example, learning_rate and subsample are the first two mentioned previously, and another technique is based on regularisation which includes two other parameters, reg_alpha (L1 regularisation based on Manhattan distance) and reg_lamda (L2 regularisation based on Euclidean distance). Both of these regularisation parameters aim to penalise XGBoost’s model complexity to make it a bit more conservative in order to reduce overfitting (Bruce, Bruce, and Gedeck 2020).
-
A full list of XGBoost classifier pipeline parameters and settings used can be retrieved as shown below. It contains a long list of parameters and one of the ways to find the optimal set of parameters is by using cross-validation (CV).
To see the default values or types of each XGBoost parameter, this XGBoost documentation link is useful (which can be cross-referenced with XGBoost’s Python API reference).
-
-
-
-
# To obtain only the parameter names for the ease of reading
-mlpipe_xgb.get_params().keys()
Some of the main XGBoost parameters that can be tuned are n_estimators, max_depth, learning_rate, subsample and reg_lambda. Here, I’m going to try to look for the best combination of learning_rate and subsample for a XGBoost classifier model for now.
-
-
# Specify parameters and distributions to be sampled
-params_dist = {
-# learning_rate usually between 0.01 - 0.1 as suggested by Raschka et al.
-# default is between 0 and 1
-"xgbclassifier__learning_rate": [0.05, 0.1, 0.3],
-# subsample default is between 0 and 1
-"xgbclassifier__subsample": [0.5, 0.7, 1.0]
-}
-
-
-
-
-
Randomised search CV
-
The following chunk of code is an example of running randomised search CV. I’ve deliberately folded the code to minimise the reading length of the post and also because the result from it is very similar to the grid search CV used below (randomised search CV run time was 13 min 33.2 seconds due to having two pipelines containing two different machine learning models for the same set of data). It’s being kept as a code reference for anyone who’d like to try it and also as an alternative way to do hyperparameter tuning.
-
-
-Code
-
## Uncomment code below to run
-# from sklearn.model_selection import RandomizedSearchCV
-# from time import time
-
-## Borrowing a utility function code from scikit_learn documentation to report best scores
-## Source: https://scikit-learn.org/stable/auto_examples/model_selection/plot_randomized_search.html#sphx-glr-auto-examples-model-selection-plot-randomized-search-py
-
-# def report(results, n_top=3):
-# for i in range(1, n_top + 1):
-# candidates = np.flatnonzero(results["rank_test_score"] == i)
-# for candidate in candidates:
-# print("Model with rank: {0}".format(i))
-# print(
-# "Mean validation score: {0:.3f} (std: {1:.3f})".format(
-# results["mean_test_score"][candidate],
-# results["std_test_score"][candidate],
-# )
-# )
-# print("Parameters: {0}".format(results["params"][candidate]))
-# print("")
-
-## The following code has also referenced and adapted from this notebook
-## https://github.com/EBjerrum/scikit-mol/blob/main/notebooks/06_hyperparameter_tuning.ipynb
-
-# n_iter_search = 9
-
-# random_search = RandomizedSearchCV(
-# mlpipe_xgb,
-# param_distributions=params_dist,
-# n_iter=n_iter_search,
-# n_jobs=2
-# )
-
-# t_start = time()
-# random_search.fit(X_train, y_train)
-# t_finish = time()
-
-# print(f'Runtime: {t_finish-t_start:0.2F} seconds for {n_iter_search} iterations')
-
-## Run report function code
-# report(random_search.cv_results_)
-
-
-
-
-
-
Grid search CV
-
-
grid_search = GridSearchCV(
- mlpipe_xgb,
- param_grid=params_dist,
- verbose=1,
- n_jobs=2
-)
-
-grid_search.fit(X_train, y_train)
-
-print(f"The best cv score is: {grid_search.best_score_:0.2f}")
-print(f"The best cv parameter settings are: {grid_search.best_params_}")
-
-# This may take longer time to run depending on computer hardware specs (for me it's taken ~13min)
-
-
Fitting 5 folds for each of 9 candidates, totalling 45 fits
-
-
-
The best cv score is: 0.99
-The best cv parameter settings are: {'xgbclassifier__learning_rate': 0.3, 'xgbclassifier__subsample': 0.7}
-
-
-
For tuning parameters of Morgan fingerprints, this Scikit_mol example notebook explains how to do it with code, so I won’t repeat them here, but have only shown how to tune some of the main XGBoost parameters.
-
-
-
-
Pickle model
-
The next step is to pickle the model or pipeline if wanting to save it for future use and to avoid re-training model from the ground up again.
One thing to remember is to avoid unpickling unknown files over insecure network, and add security key if needed.
-
-
# Pickle to save (serialise) the model in working directory (specify path if needed)
-# "wb" - write binary
-pickle.dump(mlpipe_xgb, open("xgb_pipeline.pkl", "wb"))
-# Unpickle (de-serialise) the model
-# "rb" - read binary
-mlpipe_xgb_2 = pickle.load(open("xgb_pipeline.pkl", "rb"))
-
-# Use the unpickled model object to make prediction
-pred2 = mlpipe_xgb_2.predict(X_test)
-
-## Check unpickled model and original model are the same via Python's assertion method
-#assert np.sum(np.abs(pred2 - pred)) == 0
-## or alternatively use numpy's allclose()
-print(np.allclose(pred, pred2))
-
-
True
-
-
-
-
-
-
-
Acknowledgement
-
Again, this grows into another really long post… Although this post has taken quite a long time to build up to completion, I still want to thank all the contributors or developers for all the packages used in this post.
-Chen, Tianqi, and Carlos Guestrin. 2016. “XGBoost: A Scalable Tree Boosting System.”CoRR abs/1603.02754. http://arxiv.org/abs/1603.02754.
-
-
-Freund, Yoav, and Robert E Schapire. 1997. “A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting.”Journal of Computer and System Sciences 55 (1): 119–39. https://doi.org/10.1006/jcss.1997.1504.
-
-
-Raschka, Sebastian, Yuxi (Hayden) Liu, and Vahid Mirjalili. 2022. Machine Learning with PyTorch and Scikit-Learn. Birmingham, UK: Packt Publishing.
-
]]>
- Machine learning projects
- Tree models
- Pandas
- Scikit-learn
- RDKit
- ChEMBL database
- Python
- https://jhylin.github.io/Data_in_life_blog/posts/19_ML2-3_Boosted_trees/1_adaboost_xgb.html
- Wed, 05 Jun 2024 12:00:00 GMT
-
-
- Using Molstar in Quarto
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/18_Notes_molstar_quarto/Molstar_quarto.html
-
-
Background
-
This is really a short post (note) for myself and probably for others who may be interested in software tools to visualise in silico macromolecules and small molecules.
-
Most bioinformaticians or structural biologists are probably already familiar with this software package, Molstar or Mol* (Sehnal et al. 2021). Molstar is a 3D viewer for large macromolecules (e.g. proteins), which are commonly used in structural biology and drug discovery (and also other related scientific disciplines).
-
A Quarto extension has been developed to embed the Molstar interactive 3D viewer inside Quarto markdown documents, which can be rendered as HTML pages. The main advantage of this is that it’s useful for reports or presentations.
Streamlit and Dash integrations are also possible, this also makes me think that I could probably try integrating Molstar with Shiny for Python, it’ll likely be a future side project.
-
-
-
-
-
An example using Molstar with RCSB PDB
-
The following example retrieves a protein (PDB ID: 4MQT) from RCSB PDB.
-
{{< mol-rcsb 4mqt >}}
-
-
-
-
Hover over protein structure to see details of amino acid residues or ligands present in the structure.
-
To focus or zoom-in on the ligand bound to the receptor, just click on the ligand first. This shows most of the chemical interactions between the receptor and ligand bound to it (e.g. hydrogen bondings, other chemical interactions will appear if present e.g. pi-pi stacking).
-
Screenshots or state snapshots are also available from the viewer (other utility functions can be found on the top right corner of the viewer).
-
-
-
-An close-up screenshot of 4MQT showing bound ligand
-
-
-
MD trajectories are also available, although I haven’t quite got there yet but it’s useful to know this may be possible (see example C from https://molstar.org/viewer-docs/examples/).
-
It’s also possible to upload AlphaFold-sourced proteins, or from other file sources (see examples shown from Molstar example).
-
-
-
-
-
-
References
-
-Sehnal, David, Sebastian Bittrich, Mandar Deshpande, Radka Svobodová, Karel Berka, Václav Bazgier, Sameer Velankar, Stephen K Burley, Jaroslav Koča, and Alexander S Rose. 2021. “Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures.”Nucleic Acids Research 49 (W1): W431–37. https://doi.org/10.1093/nar/gkab314.
-
-
]]>
- Notes
- Molecular viz
- https://jhylin.github.io/Data_in_life_blog/posts/18_Notes_molstar_quarto/Molstar_quarto.html
- Fri, 05 Apr 2024 11:00:00 GMT
-
-
-
- Random forest classifier
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/2_random_forest_classifier.html
- The section on “Data retrieval using chembl_downloader” has been updated and finalised on 31st January 2024 - many thanks for the comment from Charles Tapley Hoyt (cthoyt).
-
This post was really just an addition towards the last random forest (RF) post. It was mainly inspired by this paper (Esposito et al. 2021) from rinikerlab1. It was nice to complete the RF series by adding a RF classifier since last post was only on a regressor. Another thing was that imbalanced datasets were common in drug discovery projects, learning different strategies to deal with them was also very useful. While working on this post, I also came across a few other packages that I haven’t used before so I’ve included them all down below.
-
-
-
-
Overview of post
-
-
Data sourcing via chembl_downloader
-
Minor data preprocessing using own little script and also SMILES checker from scikit_mol
-
scikit-learn’s RandomForestClassifier()
-
Dealing with imbalanced dataset in RF classifiers by using ghostml
-
A small section on plotting receiver operating characteristic (ROC) curves
chembl_downloader was something I wanted to try a while back. I’ve tried manual download and chembl_webresource_client previously, and they were probably not the best strategies for data reproducibility. The idea of chembl_downloader was to generate a reproducible ChEMBL data source. It involved some SQL at the beginning to specify the exact type of data needed, so some SQL knowledge were required. Other uses for this package were elaborated much more clearly in its GitHub repository at https://github.com/cthoyt/chembl-downloader. One of the reference notebooks that I’ve used could be reached here (more available in its repository).
-
What I did was shown below.
-
-
# Show the latest version of ChEMBL used
-latest_version = latest()
-print(f"The latest ChEMBL version is: {latest_version}")
-
-
The latest ChEMBL version is: 33
-
-
-
The following section was updated as suggested by cthoyt (via his comment for post below). I ended up putting through my first ever pull request in an open-source and cheminformatics-related repository. A new option to choose max_phase was added into the get_target_sql function in chembl_downloader by keeping it as a togglable option via boolean flag. Many thanks for the patience from cthoyt for guiding me through it. The overall code was now changed as shown below.
-
::: {.cell execution_count=3}
-
# Generate SQL for a query on acetylcholinesterase (AChE): CHEMBL220
-sql = queries.get_target_sql(target_id="CHEMBL220", target_type="SINGLE PROTEIN", max_phase=True)
-
-# Pretty-print the SQL in Jupyter
-queries.markdown(sql)
-
-
SELECT
- ASSAYS.chembl_id AS assay_chembl_id,
- TARGET_DICTIONARY.target_type,
- TARGET_DICTIONARY.tax_id,
- COMPOUND_STRUCTURES.canonical_smiles,
- MOLECULE_DICTIONARY.chembl_id AS molecule_chembl_id,
- MOLECULE_DICTIONARY.max_phase,
- ACTIVITIES.standard_type,
- ACTIVITIES.pchembl_value
-FROM TARGET_DICTIONARY
-JOIN ASSAYS ON TARGET_DICTIONARY.tid == ASSAYS.tid
-JOIN ACTIVITIES ON ASSAYS.assay_id == ACTIVITIES.assay_id
-JOIN MOLECULE_DICTIONARY ON MOLECULE_DICTIONARY.molregno == ACTIVITIES.molregno
-JOIN COMPOUND_STRUCTURES ON MOLECULE_DICTIONARY.molregno == COMPOUND_STRUCTURES.molregno
-WHERE TARGET_DICTIONARY.chembl_id ='CHEMBL220'
-AND ACTIVITIES.pchembl_value ISNOTNULL
-AND TARGET_DICTIONARY.target_type ='SINGLE PROTEIN'```
-:::
-:::
-
-
-::: {.cell execution_count=4}
-``` {.python .cell-code}
-# The SQL still works as shown above,
-# please ignore the non-SQL texts
-# - bit of a formatting issue
-# when pretty-printing the sql,
-# but shouldn't affect the code
-
-
I’ve also updated how I retrieved and saved the ChEMBL data with the following code suggested and provided by cthoyt. This would be a better and more reproducible way for anyone who might be interested in re-running this notebook.
-
-
from pathlib import Path
-
-# Pick any directory, but make sure it's relative to your home directory
-directory = Path.home().joinpath(".data", "blog")
-# Create the directory if it doesn't exist
-directory.mkdir(exist_ok=True, parents=True)
-
-# Create a file path that corresponds to the version, since this could change
-path = directory.joinpath(f"chembl_d_ache_{latest_version}.tsv")
-
-if path.is_file():
-# If the file already exists, load it
- df_ache = pd.read_csv(path, sep=',')
-else:
-# If the file doesn't already exist, make the query then cache it
- df_ache = chembl_downloader.query(sql)
- df_ache.to_csv(path, sep=",", index=False)
-
-
The rest of the code outputs in the post stayed the same as before. The only thing changed and updated was the part on retrieving ChEMBL data via chembl_downloader.
-
-
-
-
Some data cleaning
-
Minor cleaning and preprocessing were done for this post only, as the focus was more on dealing with imbalanced dataset in RF classifier. Since I used a different way to retrieve ChEMBL data this time, the dataset used here might be slightly different from the one used in previous post.
-
-
-
mol_prep.py
-
I’ve more or less accumulated small pieces of code over time, and I’ve decided to compile them into a Python script. The idea was to remove most function code in the post to avoid repeating them all the time since they’ve been used frequently in the last few posts. The script would be saved into the RF repository, and would still be considered as a “work-in-progress” script (needs more work in the future).
-
-
## Trial own mol_prep.py script
-from mol_prep import preprocess, rdkit_2d_descriptors
scikit_mol was a package originated from RDKit UGM hackathon in 2022. This blog post elaborated further on its functions and uses in machine learning. For this post I’ve only used it for a very small portion, mainly to check for missing SMILES or errors in SMILES (kind of like double checking whether the preprocess function code worked as expected). It could be integrated with scikit-learn’s pipeline method for multiple estimators. Its GitHub Repository link: https://github.com/EBjerrum/scikit-mol - I’ve referred to this reference notebook while working on this post.
-
-
# Quick simple way to check for missing SMILES
-print(f'Dataset contains {df_prep.standard_smiles.isna().sum()} unparsable mols')
-
-
Dataset contains 0 unparsable mols
-
-
-
-
# Checking for invalid SMILES using scikit_mol
-smileschecker = CheckSmilesSanitazion()
-smileschecker.sanitize(list(df_prep.standard_smiles))
-
-# Showing SMILES errors
-smileschecker.errors
-
-
-
-
-
-
-
-
-
SMILES
-
-
-
-
-
-
-
-
-
It showed no errors in SMILES (errors should be listed in the code cell output).
A different spreads of max phases were shown this time in the dataframe, as the SQL query mainly used IC50, whereas last post was strictly limited to Ki via ChEMBL web resource client. Other likely reason was that in the decision tree series, I’ve attempted data preprocessing at a larger scale so some data were eliminated. It appeared that there were more max phase 4 compounds here than last time (note: null compounds were not shown in the value counts as it was labelled as “NaN”, it should be the largest max phase portion in the data).
-
-
# Find out counts of each max phase
-df_merge.value_counts("max_phase")
# Dropping duplicated compound via chembl IDs in the main df
-df_merge_new = df_merge.drop_duplicates(subset=["molecule_chembl_id"], keep="first")
-print(df_merge_new.shape)
-df_merge_new.head()
-
-
(5357, 212)
-
-
-
-
-
-
-
-
-
-
max_phase
-
molecule_chembl_id
-
MaxAbsEStateIndex
-
MaxEStateIndex
-
MinAbsEStateIndex
-
MinEStateIndex
-
qed
-
SPS
-
MolWt
-
HeavyAtomMolWt
-
...
-
fr_sulfide
-
fr_sulfonamd
-
fr_sulfone
-
fr_term_acetylene
-
fr_tetrazole
-
fr_thiazole
-
fr_thiocyan
-
fr_thiophene
-
fr_unbrch_alkane
-
fr_urea
-
-
-
-
-
0
-
4.0
-
CHEMBL411
-
9.410680
-
9.410680
-
0.284153
-
0.284153
-
0.779698
-
12.100000
-
268.356
-
248.196
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
1
-
4.0
-
CHEMBL416
-
11.173100
-
11.173100
-
0.405828
-
-0.405828
-
0.586359
-
11.062500
-
216.192
-
208.128
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
3
-
-1.0
-
CHEMBL7002
-
11.591481
-
11.591481
-
0.189306
-
-0.309798
-
0.886859
-
23.608696
-
333.453
-
310.269
-
...
-
1
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
4
-
NaN
-
CHEMBL28
-
12.020910
-
12.020910
-
0.018823
-
-0.410347
-
0.631833
-
10.800000
-
270.240
-
260.160
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
5
-
4.0
-
CHEMBL41
-
12.564531
-
12.564531
-
0.203346
-
-4.329869
-
0.851796
-
12.909091
-
309.331
-
291.187
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
-
5 rows × 212 columns
-
-
-
-
-
# Making sure previously used 10 max phase 4 compounds could be found in df_merge_new
-df_mp4 = df_merge_new.loc[df_merge_new["molecule_chembl_id"].isin(list_mp4)]
-df_mp4
-
-
-
-
-
-
-
-
-
max_phase
-
molecule_chembl_id
-
MaxAbsEStateIndex
-
MaxEStateIndex
-
MinAbsEStateIndex
-
MinEStateIndex
-
qed
-
SPS
-
MolWt
-
HeavyAtomMolWt
-
...
-
fr_sulfide
-
fr_sulfonamd
-
fr_sulfone
-
fr_term_acetylene
-
fr_tetrazole
-
fr_thiazole
-
fr_thiocyan
-
fr_thiophene
-
fr_unbrch_alkane
-
fr_urea
-
-
-
-
-
9
-
4.0
-
CHEMBL481
-
13.581173
-
13.581173
-
0.095133
-
-1.863974
-
0.355956
-
22.209302
-
586.689
-
548.385
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
67
-
4.0
-
CHEMBL95
-
6.199769
-
6.199769
-
0.953981
-
0.953981
-
0.706488
-
15.200000
-
198.269
-
184.157
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
249
-
4.0
-
CHEMBL502
-
12.936933
-
12.936933
-
0.108783
-
0.108783
-
0.747461
-
20.214286
-
379.500
-
350.268
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
508
-
4.0
-
CHEMBL640
-
11.743677
-
11.743677
-
0.044300
-
-0.044300
-
0.731540
-
10.529412
-
235.331
-
214.163
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
512
-
4.0
-
CHEMBL659
-
9.972866
-
9.972866
-
0.008380
-
-0.411699
-
0.800524
-
33.857143
-
287.359
-
266.191
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
1013
-
4.0
-
CHEMBL1025
-
12.703056
-
12.703056
-
0.426312
-
-4.304784
-
0.629869
-
13.000000
-
184.147
-
170.035
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
1345
-
4.0
-
CHEMBL1128
-
9.261910
-
9.261910
-
0.000000
-
0.000000
-
0.608112
-
10.692308
-
201.697
-
185.569
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
2028
-
4.0
-
CHEMBL360055
-
6.476818
-
6.476818
-
0.656759
-
0.656759
-
0.205822
-
12.583333
-
510.828
-
450.348
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
2725
-
4.0
-
CHEMBL1677
-
6.199769
-
6.199769
-
0.000000
-
0.000000
-
0.760853
-
14.250000
-
234.730
-
219.610
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
3271
-
4.0
-
CHEMBL1200970
-
2.520809
-
2.520809
-
0.000000
-
0.000000
-
0.709785
-
14.000000
-
348.943
-
323.743
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
-
10 rows × 212 columns
-
-
-
-
-
# note: compounds with max phase 0 not shown in the count
-df_merge_new.value_counts("max_phase")
The aim of this post was to model and classify the max phases of ChEMBL small molecules, i.e. whether the compounds in the testing set (consisted of max phase 0 or null compounds) might be eventually classified as max phase 4 or not. This was one of the approaches to answer the question in mind, and not the ultimate way to solve the problem (just thought to mention). The target was “max_phase” and features to be used were the various RDKit 2D descriptors (RDKit2D).
-
The steps I’ve taken to build the model were shown below:
-
-
Re-labelled max phases as binary labels (e.g. max phase null as 0, max phase 4 as 1)
-
-
-
# Re-label max phase NaN as 0
-df_merge_new = df_merge_new.fillna(0)
-df_merge_new.head()
-
-
-
-
-
-
-
-
-
max_phase
-
molecule_chembl_id
-
MaxAbsEStateIndex
-
MaxEStateIndex
-
MinAbsEStateIndex
-
MinEStateIndex
-
qed
-
SPS
-
MolWt
-
HeavyAtomMolWt
-
...
-
fr_sulfide
-
fr_sulfonamd
-
fr_sulfone
-
fr_term_acetylene
-
fr_tetrazole
-
fr_thiazole
-
fr_thiocyan
-
fr_thiophene
-
fr_unbrch_alkane
-
fr_urea
-
-
-
-
-
0
-
4.0
-
CHEMBL411
-
9.410680
-
9.410680
-
0.284153
-
0.284153
-
0.779698
-
12.100000
-
268.356
-
248.196
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
1
-
4.0
-
CHEMBL416
-
11.173100
-
11.173100
-
0.405828
-
-0.405828
-
0.586359
-
11.062500
-
216.192
-
208.128
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
3
-
-1.0
-
CHEMBL7002
-
11.591481
-
11.591481
-
0.189306
-
-0.309798
-
0.886859
-
23.608696
-
333.453
-
310.269
-
...
-
1
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
4
-
0.0
-
CHEMBL28
-
12.020910
-
12.020910
-
0.018823
-
-0.410347
-
0.631833
-
10.800000
-
270.240
-
260.160
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
5
-
4.0
-
CHEMBL41
-
12.564531
-
12.564531
-
0.203346
-
-4.329869
-
0.851796
-
12.909091
-
309.331
-
291.187
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
-
5 rows × 212 columns
-
-
-
-
-
Splitted data into max phase null & max phase 4 (needing to re-label max phase 4 column only as 1, and not disrupting the labels of max phase 0 compounds)
-
-
-
# Select all max phase null compounds
-df_null = df_merge_new[df_merge_new["max_phase"] ==0]
-print(df_null.shape)
-df_null.head()
-
-
(5256, 212)
-
-
-
-
-
-
-
-
-
-
max_phase
-
molecule_chembl_id
-
MaxAbsEStateIndex
-
MaxEStateIndex
-
MinAbsEStateIndex
-
MinEStateIndex
-
qed
-
SPS
-
MolWt
-
HeavyAtomMolWt
-
...
-
fr_sulfide
-
fr_sulfonamd
-
fr_sulfone
-
fr_term_acetylene
-
fr_tetrazole
-
fr_thiazole
-
fr_thiocyan
-
fr_thiophene
-
fr_unbrch_alkane
-
fr_urea
-
-
-
-
-
4
-
0.0
-
CHEMBL28
-
12.020910
-
12.020910
-
0.018823
-
-0.410347
-
0.631833
-
10.800000
-
270.240
-
260.160
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
8
-
0.0
-
CHEMBL8320
-
10.282778
-
10.282778
-
0.120741
-
-0.120741
-
0.416681
-
17.500000
-
108.096
-
104.064
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
14
-
0.0
-
CHEMBL11833
-
11.201531
-
11.201531
-
0.428520
-
-0.466092
-
0.838024
-
25.157895
-
262.309
-
244.165
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
392
-
0.0
-
CHEMBL12324
-
11.257704
-
11.257704
-
0.462395
-
-0.462395
-
0.797990
-
26.150000
-
277.344
-
256.176
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
393
-
0.0
-
CHEMBL274107
-
11.359778
-
11.359778
-
0.372211
-
-0.473241
-
0.838024
-
25.157895
-
262.309
-
244.165
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
-
5 rows × 212 columns
-
-
-
-
-
# Using pd.DataFrame.assign to add a new column to re-label max_phase 4 as "1"
-df_mp4_lb = df_mp4.assign(max_phase_lb = df_mp4["max_phase"] /4)
-
-# Using pd.DataFrame.pop() & insert() to shift added column to first column position
-first_col = df_mp4_lb.pop("max_phase_lb")
-df_mp4_lb.insert(0, "max_phase_lb", first_col)
-df_mp4_lb.head()
-
-
-
-
-
-
-
-
-
max_phase_lb
-
max_phase
-
molecule_chembl_id
-
MaxAbsEStateIndex
-
MaxEStateIndex
-
MinAbsEStateIndex
-
MinEStateIndex
-
qed
-
SPS
-
MolWt
-
...
-
fr_sulfide
-
fr_sulfonamd
-
fr_sulfone
-
fr_term_acetylene
-
fr_tetrazole
-
fr_thiazole
-
fr_thiocyan
-
fr_thiophene
-
fr_unbrch_alkane
-
fr_urea
-
-
-
-
-
9
-
1.0
-
4.0
-
CHEMBL481
-
13.581173
-
13.581173
-
0.095133
-
-1.863974
-
0.355956
-
22.209302
-
586.689
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
67
-
1.0
-
4.0
-
CHEMBL95
-
6.199769
-
6.199769
-
0.953981
-
0.953981
-
0.706488
-
15.200000
-
198.269
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
249
-
1.0
-
4.0
-
CHEMBL502
-
12.936933
-
12.936933
-
0.108783
-
0.108783
-
0.747461
-
20.214286
-
379.500
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
508
-
1.0
-
4.0
-
CHEMBL640
-
11.743677
-
11.743677
-
0.044300
-
-0.044300
-
0.731540
-
10.529412
-
235.331
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
512
-
1.0
-
4.0
-
CHEMBL659
-
9.972866
-
9.972866
-
0.008380
-
-0.411699
-
0.800524
-
33.857143
-
287.359
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
-
5 rows × 213 columns
-
-
-
-
-
# Also create a new column max_phase_lb column for df_null
-# in order to merge 2 dfs later
-df_null_lb = df_null.assign(max_phase_lb = df_null["max_phase"])
-first_col_null = df_null_lb.pop("max_phase_lb")
-df_null_lb.insert(0, "max_phase_lb", first_col_null)
-df_null_lb.head()
# Convert both X & y to arrays
-X = X.to_numpy()
-y = y.to_numpy()
-
-
-
# Using train_test_split() this time to split data
-X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, stratify=y, random_state=1)
-
-
After data splitting, a RF classifier was trained with reference to this notebook.
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Extracted positive prediction probabilities for the testing set and showed confusion matrix with classification metrics
-
-
-
test_probs = rfc.predict_proba(X_test)[:, 1]
-
-
Some reference links and explanations for area under the ROC curve and Cohen’s Kappa.
-
Area under the ROC curve: reference - the area under a curve plot between sensitivity or recall (percent of all 1s classified correctly by a classifier or true positive rate) and specificity (percent of all 0s classified correctly by a classifier, or equivalent to 1 - false positive rate or true negative rate) (Bruce, Bruce, and Gedeck 2020). It is useful for evaluating the performance of a classification model via comparing the true positive rate and false positive rate which are influenced by shifting the decision threshold. Area under the ROC is usually represented as a number ranging from 0 to 1 (1 being a perfect classifier, 0.5 or below meaning a poor, ineffective classifier)
-
Cohen’s Kappa score: reference - a score that is used to measure the agreement of labelling between two annotators (usually between -1 and 1, the higher the score the better the agreement)
-
Rather than re-inventing the wheel, the following function code for calculating metrics of the RF model were adapted from this notebook, from GHOST repository. I have only added some comments for clarities, and also added a zero_division parameter for the classification_report to mute the warning message when the results ended up being 0 due to divisions by zero.
-
-
def calc_metrics(y_test, test_probs, threshold =0.5):
-# Target label assigned according to stated decision threshold (default = 0.5)
-# e.g. second annotator (expected label)
- scores = [1if x>=threshold else0for x in test_probs]
-# Calculate area under the ROC curve based on prediction score
- auc = metrics.roc_auc_score(y_test, test_probs)
-# Calculate Cohen's Kappa score
-# e.g. y_test as first annotator (predicted label)
- kappa = metrics.cohen_kappa_score(y_test, scores)
-# Formulate the confusion matrix
- confusion = metrics.confusion_matrix(y_test, scores, labels =list(set(y_test)))
-print('thresh: %.2f, kappa: %.3f, AUC test-set: %.3f'%(threshold, kappa, auc))
-print(confusion)
-print(metrics.classification_report(y_test, scores, zero_division=0.0))
-return
-
-
Note: roc_auc_score measures true positive and false positive rates, requiring binary labels (e.g. 0s and 1s) in the data
-
Then showed confusion matrix along with area under the ROC curve and Cohen’s Kappa.
It was very obvious that not all of the compounds were classified in the testing set. There were only 1052 compounds classified as true negative, and none in the testing set were labelled as true positive. The likely reason was due to the very imbalanced ratio of actives (only 10 max phase 4 which were labelled as “1” compounds) and inactives (5256 max phase 0 compounds). Besides the imbalanced dataset, the decision threshold was also normally set at 0.5, meaning the classifier was likely going to lose the chance to classify the true positive compounds due to the very skewed ratio of actives to inactives.
-
-
Two approaches were used in the GHOST (generalized threshold shifting) paper:
Approach 2 led to GHOST procedure with a goal to optimise and shift the decision threshold in any classification methods to catch the minor portion of actives (rather than the major portion of inactives)
-
note: both approaches were shown to be performing similarly in the paper
-
-
-
I only used approach 2 here since the RDKit blog post had already explained approach 1 in depth.
-
The next step involved extracting prediction probabilities from the RF classifier trained model.
-
-
# Get the positive prediction probabilities of the training set
-train_probs = rfc.predict_proba(X_train)[:, 1]
-
-
-
Used GHOST strategy in a postprocessing way (note: last post used data re-sampling method in a preprocessing way)
-
-
The decision threshold were optimised by using ghostml code via testing various different thresholds, e.g. in spaces of 0.05 that ranged from 0.05 to 0.5. The most optimal threshold would have the most maximised Cohen’s kappa.
-
-
# Setting up different decision thresholds
-thresholds = np.round(np.arange(0.05,0.55,0.05), 2)
-thresholds
# Looking for the best threshold with the most optimal Cohen's Kappa
-new_threshold = ghostml.optimize_threshold_from_predictions(y_train, train_probs, thresholds, ThOpt_metrics ='ROC')
-
-
Using the calc_metrics function again on the newly-found or shifted decision threshold.
Here, after shifting the decision threshold with the most optimal Cohen’s Kappa score, we could see an improved number of compounds labelled within the true negative class (increasing from 1052 to 4204), and more importantly, we could see the true positive class improved from 0 to 7 as well.
-
-
-
-
Plotting ROC curves
-
Time for some plots - I’ve shown two different ways to plot ROC curves below.
-
-
Using scikit-learn
-
-
Testing set ROC curve - obviously, this was not a good classifier with a poor AUC.
I wanted to mention that the testing set used here was most likely not the best ones to be used. There could be many overlaps or similarities between the training and testing sets, since they all came from ChEMBL database. For demonstration and learning purposes, I ended up using similar dataset as last time. Hopefully, I can try other open-source or public drug discovery datasets in the near future.
-
The other thing to mention was that I should try different molecular fingerprints or descriptors as well, rather than only using RDKit2D, which might lead to different results. I should also probably slowly move onto using multiple datasets or targets in a project, which would likely make things more interesting. On the other hand, I also wanted to avoid this in order to make the topic of interest as clear and simple as possible for me or anyone who’s trying to learn.
-
-
-
-
Acknowledgements
-
I’d like to thank Riniker lab again for the GHOST paper, along with all the authors, contributors or developers for all of the software packages mentioned in this post, and also, huge thanks should also go to the authors of the reference notebooks mentioned in the post as well.
-Esposito, Carmen, Gregory A. Landrum, Nadine Schneider, Nikolaus Stiefl, and Sereina Riniker. 2021. “GHOST: Adjusting the Decision Threshold to Handle Imbalanced Data in Machine Learning.”Journal of Chemical Information and Modeling 61 (6): 2623–40. https://doi.org/10.1021/acs.jcim.1c00160.
-
-
Footnotes
-
-
-
h/t: Greg Landrum for his comment on Mastodon for the last RF post (which led to this follow-up post)↩︎
-
-
]]>
- Machine learning projects
- Tree models
- Pandas
- Scikit-learn
- RDKit
- ChEMBL database
- Python
- https://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/2_random_forest_classifier.html
- Tue, 16 Jan 2024 11:00:00 GMT
-
-
- Random forest
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/1_random_forest.html
- Post updated on 3rd May 2024 - Added comment regarding ImbalancedLearningRegression package (installation tip) & Jupyter notebook link of this post
-
-
Quick overview of this post
-
-
Short introduction of random forest
-
Random forest methods or classes in scikit-learn
-
Random forest regressor model in scikit-learn
-
Training and testing data splits
-
-
ChEMBL-assigned max phase splits
-
Imbalanced learning regression and max phase splits
-
-
Scoring metrics of trained models
-
Feature importances in dataset
-
-
feature_importances_attribute in scikit-learn
-
permutation_importance function in scikit-learn
-
SHAP approach
-
-
Hyperparameter tuning on number of trees
-
-
-
-
-
What is a random forest?
-
The decision tree model built last time was purely based on one model on its own, which often might not be as accurate or reflective in real-life. To improve the model, the average outcome from multiple models (Breiman 1998) should be considered to see if this would provide a more realistic image. This model averaging approach was also constantly used in our daily lives, for example, using majority votes during decision-making steps.
-
The same model averaging concept was also used in random forest (Breiman 2001), which as the name suggested, was composed of many decision trees (models) forming a forest. Each tree model would be making its own model prediction. By accruing multiple predictions since we have multiple trees, the average obtained from these predictions would produce one single result in the end. The advantage of this was that it improved the accuracy of the prediction by reducing variances, and also minimised the problem of overfitting the model if it was purely based on one model only (more details in section 1.11.2.1. Random Forests from scikit-learn).
-
The “random” part of the random forest was introduced in two ways. The first one was via using bootstrap samples, which was also known as bagging or bootstrap aggregating (Bruce, Bruce, and Gedeck 2020), where samples were drawn with replacements within the training datasets for each tree built in the ensemble (also known as the perturb-and-combine technique (Breiman 1998)). While bootstrap sampling was happening, randomness was also incorporated into the training sets at the same time. The second way randomness was introduced was by using a random subset of features for splitting at the nodes, or a full set of features could also be used (although this was generally not recommended). The main goal here was to achieve best splits at each node.
-
-
-
-
Random forest in scikit-learn
-
Scikit-learn had two main types of random forest classes - ensemble.RandomForestClassifier() and ensemble.RandomForestRegressor(). When to use which class would depend on the target values. The easiest thing to do was to decide whether the target variables had class labels (binary types or non-continuous variables e.g. yes or no, or other different categories to be assigned) or continuous (numerical) variables, which in this case, if I were to continue using the same dataset from the decision tree series, it would be a continuous variable or feature, pKi, the inhibition constant.
-
There were also two other alternative random forest methods in scikit-learn, which were ensemble.RandomTreesEmbedding() and ensemble.ExtraTreesClassifier() or ensemble.ExtraTreesRegressor(). The difference for RandomTreesEmbedding() was that it was an unsupervised method that used data transformations (more details from section 1.11.2.6. on “Totally Random Trees Embedding” in scikit-learn). On the other side, there was also an option to use ExtraTreesClassifier() or ExtraTreesRegressor() to generate extremely randomised trees that would go for another level up in randomness (more deatils in section 1.11.2.2. on Extremely Randomized Trees from scikit-learn). The main difference for this type of random forest was that while there was already a random subset of feature selection used (with an intention to select the most discerning features), more randomness were added on top of this by using purely randomly generated splitting rules for picking features at the nodes. The advantage of this type of method was that it would reduce variance and increase the accuracy of the model, but the downside was there might be an increase in bias within the model.
-
-
-
-
Building a random forest regressor model using scikit-learn
-
As usual, all the required libraries were imported first.
-
-
import pandas as pd
-import sklearn
-from sklearn.ensemble import RandomForestRegressor
-
-# For imbalanced datasets in regression
-# May need to set env variable (SKLEARN_ALLOW_DEPRECATED_SKLEARN_PACKAGE_INSTALL=True) when installing
-# due to package dependency on older sklearn version
-import ImbalancedLearningRegression as iblr
-
-# Plots
-import matplotlib.pyplot as plt
-import seaborn as sns
-
-# Metrics
-from sklearn.metrics import mean_squared_error
-from sklearn.metrics import r2_score
-
-# Feature importances
-# Permutation_importance
-from sklearn.inspection import permutation_importance
-# SHAP values
-import shap
-
-# Hyperparameter tuning
-from sklearn.model_selection import cross_val_score, RepeatedKFold
-
-from numpy import mean, std
-from natsort import index_natsorted
-import numpy as np
-
-# Showing version of scikit-learn used
-print(sklearn.__version__)
-
-
1.3.2
-
-
-
Importing dataset that was preprocessed from last time - link to data source: first decision tree post.
-
-
data = pd.read_csv("ache_2d_chembl.csv")
-data.drop(columns = ["Unnamed: 0"], inplace=True)
-# Preparing data for compounds with max phase with "NaN" by re-labelling to "null"
-data["max_phase"].fillna("null", inplace=True)
-data.head()
-
-
Setting an item of incompatible dtype is deprecated and will raise in a future error of pandas. Value 'null' has dtype incompatible with float64, please explicitly cast to a compatible dtype first.
-
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
pKi
-
max_phase
-
mw
-
fsp3
-
n_lipinski_hba
-
n_lipinski_hbd
-
n_rings
-
n_hetero_atoms
-
n_heavy_atoms
-
...
-
sas
-
n_aliphatic_carbocycles
-
n_aliphatic_heterocyles
-
n_aliphatic_rings
-
n_aromatic_carbocycles
-
n_aromatic_heterocyles
-
n_aromatic_rings
-
n_saturated_carbocycles
-
n_saturated_heterocyles
-
n_saturated_rings
-
-
-
-
-
0
-
CHEMBL60745
-
8.787812
-
null
-
245.041526
-
0.400000
-
2
-
1
-
1
-
3
-
13
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
1
-
CHEMBL208599
-
10.585027
-
null
-
298.123676
-
0.388889
-
2
-
2
-
4
-
3
-
21
-
...
-
4.331775
-
2
-
0
-
2
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
2
-
CHEMBL95
-
6.821023
-
4.0
-
198.115698
-
0.307692
-
2
-
2
-
3
-
2
-
15
-
...
-
2.014719
-
1
-
0
-
1
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
3
-
CHEMBL173309
-
7.913640
-
null
-
694.539707
-
0.666667
-
8
-
0
-
2
-
8
-
50
-
...
-
2.803680
-
0
-
0
-
0
-
2
-
0
-
2
-
0
-
0
-
0
-
-
-
4
-
CHEMBL1128
-
6.698970
-
4.0
-
201.092042
-
0.400000
-
2
-
1
-
1
-
3
-
13
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
-
5 rows × 25 columns
-
-
-
-
-
-
-
Training/testing splits
-
Two approaches were used, where one was based purely on max phase split (between max phases null and 4), which was used last time in the decision tree series, and the other one was using the same max phase split but with an ImbalancedLearningRegression method added on top of it.
-
-
-
Preparing training data using max phase split
-
X variable was set up first from the dataframe, and then converted into a NumPy array, which consisted of the number of samples and number of features. This was kept the same as how it was in the decision tree posts.
-
-
-
-
-
-
-Note
-
-
-
-
It’s usually recommended to copy the original data or dataframe before doing any data manipulations to avoid unnecessary changes to the original dataset (this was not used in the decision tree posts, but since I’m going to use the same set of data again I’m doing it here.)
-
-
-
-
# X variables (molecular features)
-# Make a copy of the original dataframe first
-data_mp4 = data.copy()
-# Selecting all max phase 4 compounds
-data_mp4 = data_mp4[data_mp4["max_phase"] ==4]
-print(data_mp4.shape)
-data_mp4.head()
Again, y variable was arranged via the dataframe as well, and converted into a NumPy array. It consisted of the number of samples only as this was the target variable.
Both X and y variables were used to fit the RandomForestRegressor() estimator.
-
-
# n_estimators = 100 by default
-# note: if wanting to use whole dataset - switch off "bootstrap" parameter by using "False"
-rfreg = RandomForestRegressor(max_depth=3, random_state=1, max_features=0.3)
-rfreg.fit(X_mp4, y_mp4)
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Testing data was mainly based on compounds with max phase assigned as “0” or “null” after I renamed it above.
-
-
data_mp_null = data.copy()
-# Selecting all max phase "null" compounds
-data_mp_null = data_mp_null[data_mp_null["max_phase"] =="null"]
-print(data_mp_null.shape)
-data_mp_null.head()
-
-
(466, 25)
-
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
pKi
-
max_phase
-
mw
-
fsp3
-
n_lipinski_hba
-
n_lipinski_hbd
-
n_rings
-
n_hetero_atoms
-
n_heavy_atoms
-
...
-
sas
-
n_aliphatic_carbocycles
-
n_aliphatic_heterocyles
-
n_aliphatic_rings
-
n_aromatic_carbocycles
-
n_aromatic_heterocyles
-
n_aromatic_rings
-
n_saturated_carbocycles
-
n_saturated_heterocyles
-
n_saturated_rings
-
-
-
-
-
0
-
CHEMBL60745
-
8.787812
-
null
-
245.041526
-
0.400000
-
2
-
1
-
1
-
3
-
13
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
1
-
CHEMBL208599
-
10.585027
-
null
-
298.123676
-
0.388889
-
2
-
2
-
4
-
3
-
21
-
...
-
4.331775
-
2
-
0
-
2
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
3
-
CHEMBL173309
-
7.913640
-
null
-
694.539707
-
0.666667
-
8
-
0
-
2
-
8
-
50
-
...
-
2.803680
-
0
-
0
-
0
-
2
-
0
-
2
-
0
-
0
-
0
-
-
-
5
-
CHEMBL102226
-
4.698970
-
null
-
297.152928
-
0.923077
-
3
-
0
-
0
-
5
-
18
-
...
-
2.965170
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
7
-
CHEMBL103873
-
5.698970
-
null
-
269.121628
-
0.909091
-
3
-
0
-
0
-
5
-
16
-
...
-
3.097106
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
-
5 rows × 25 columns
-
-
-
-
-
# Set up X test variable with the same molecular features
-X_mp_test_df = data_mp_null[['mw', 'fsp3', 'n_lipinski_hba', 'n_lipinski_hbd', 'n_rings', 'n_hetero_atoms', 'n_heavy_atoms', 'n_rotatable_bonds', 'n_radical_electrons', 'tpsa', 'qed', 'clogp', 'sas', 'n_aliphatic_carbocycles', 'n_aliphatic_heterocyles', 'n_aliphatic_rings', 'n_aromatic_carbocycles', 'n_aromatic_heterocyles', 'n_aromatic_rings', 'n_saturated_carbocycles', 'n_saturated_heterocyles', 'n_saturated_rings']]
-
-# Convert X test variables from df to arrays
-X_mp_test = X_mp_test_df.to_numpy()
-
-X_mp_test
Training/testing splits using ImbalancedLearningRegression and max phase splits
-
I didn’t really pay a lot of attentions when I was doing data splits in the decision tree series, as my main focus was on building a single tree in order to fully understand and see what could be derived from just one tree. Now, when I reached this series on random forest, I realised I forgot to mention in the last series that data splitting was actually very crucial on model performance and could influence outcome predictions. It could also become quite complicated as more approaches were available to split the data. Also, the way the data was splitted could produce different outcomes.
-
After I’ve splitted the same dataset based on compounds’ max phase assignments in ChEMBL and also fitted the training data on the random forest regressor, I went back and noticed that the training and testing data were very imbalanced and I probably should do something about it before fitting them onto another model.
-
At this stage, I went further to look into whether imbalanced datasets should be addressed in regression tasks, and did a surface search online. So based on common ML concensus, addressing imbalanced datasets were more applicable to classification tasks (e.g. binary labels or multi-class labels), rather than regression problems. However, recent ML research looked into the issue of imbalanced datasets in regression. This blog post mentioned a few studies that looked into this type of problem, and I thought they were very interesting and worth a mention at least. One of them that I’ve looked into was SMOTER, which was based on synthetic minority over-sampling technique (SMOTE)(Chawla et al. 2002), and was named this way because it was basically a SMOTE for regression (hence SMOTER)(Torgo et al. 2013). Synthetic minority over-sampling technique for regression with Gaussian noise (SMOGN)(Kunz 2020) was another technique that was built upon SMOTER, but with Gaussian noises added. This has subsequently led me to ImbalancedLearningRegression library (Wu, Kunz, and Branco 2022), which was a variation of SMOGN. This was the one used on my imbalanced dataset, shown in the section below.
-
A simple flow diagram was drawn below showing the evolution of different techniques when dealing with imbalanced datasets in classification (SMOTE) and regression (SMOTER, SMOGN and ImbalancedLearningRegression):
-
-
-
-
-
flowchart LR
- A(SMOTE) --> B(SMOTER)
- B --> C(SMOGN)
- C --> D(ImbalancedLearningRegression)
-
-
-
-
-
-
-
-
-
GitHub repository for ImbalancedLearningRegression package is available here, with its documentation available here.
-
Also, I just wanted to mention that these were not the only techniques available for treating imbalanced datasets in regression, as there were other ones in the literature and most likely more are being developed currently, but I only had time to cover these here for now.
-
I also would like to mention another really useful open-source resource for treating imbalanced datasets in classifications since I did not use it in this post due to the problem being more of a regression one than a classification one - imbalance-learn library.
-
-
# Original dataset - checking shape again
-print(data.shape)
-data.head()
-
-
(481, 25)
-
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
pKi
-
max_phase
-
mw
-
fsp3
-
n_lipinski_hba
-
n_lipinski_hbd
-
n_rings
-
n_hetero_atoms
-
n_heavy_atoms
-
...
-
sas
-
n_aliphatic_carbocycles
-
n_aliphatic_heterocyles
-
n_aliphatic_rings
-
n_aromatic_carbocycles
-
n_aromatic_heterocyles
-
n_aromatic_rings
-
n_saturated_carbocycles
-
n_saturated_heterocyles
-
n_saturated_rings
-
-
-
-
-
0
-
CHEMBL60745
-
8.787812
-
null
-
245.041526
-
0.400000
-
2
-
1
-
1
-
3
-
13
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
1
-
CHEMBL208599
-
10.585027
-
null
-
298.123676
-
0.388889
-
2
-
2
-
4
-
3
-
21
-
...
-
4.331775
-
2
-
0
-
2
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
2
-
CHEMBL95
-
6.821023
-
4.0
-
198.115698
-
0.307692
-
2
-
2
-
3
-
2
-
15
-
...
-
2.014719
-
1
-
0
-
1
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
3
-
CHEMBL173309
-
7.913640
-
null
-
694.539707
-
0.666667
-
8
-
0
-
2
-
8
-
50
-
...
-
2.803680
-
0
-
0
-
0
-
2
-
0
-
2
-
0
-
0
-
0
-
-
-
4
-
CHEMBL1128
-
6.698970
-
4.0
-
201.092042
-
0.400000
-
2
-
1
-
1
-
3
-
13
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
-
5 rows × 25 columns
-
-
-
-
So my little test on using ImbalancedLearningRegression package started from below.
-
-
iblr_data = data.copy()
-
-# Introducing Gaussian noise for data sampling
-data_gn = iblr.gn(data = iblr_data, y ="pKi", pert =1)
-print(data_gn.shape)
# Followed by max phase split, where max phase 4 = training dataset
-data_gn_mp4 = data_gn[data_gn["max_phase"] ==4]
-data_gn_mp4
-print(data_gn_mp4.shape)
-
-
(7, 25)
-
-
-
-
# Also splitted max phase null compounds = testing dataset
-data_gn_mp_null = data_gn[data_gn["max_phase"] =="null"]
-data_gn_mp_null
-print(data_gn_mp_null.shape)
-
-
(465, 25)
-
-
-
There were several different sampling techniques in ImbalancedLearningRegression package. I’ve only tried random over-sampling, under-sampling and Gaussian noise, but there were also other ones such as SMOTE and ADASYN (in over-sampling technique) or condensed nearest neighbor, Tomeklinks and edited nearest neightbour (in under-sampling technique) that I haven’t used.
-
Random over-sampling actually oversampled the max phase null compounds (sample size increased), while keeping all 10 max phase 4 compounds. Under-sampling removed all of the max phase 4 compounds (which was most likely not the best option, since I was aiming to use them as training compounds), with max phase null compounds also reduced in size too. Due to post length, I did not show the code for random over-sampling and under-sampling, but for people who are interested, I think it would be interesting to test them out.
-
I ended up using Gauissian noise sampling and it reduced max phase 4 compounds slightly, and increased the max phase null compounds a little bit too, which seemed to be the most balanced data sampling at the first try. (Note: as stated from the documentation for ImbalancedLearningRegression package, missing values within features would be removed automatically, I’ve taken care of this in my last series of posts so no difference were observed here.)
-
The change in the distribution of pKi values for the Gaussian noise sampling method between the original and sample-modified datasets could be seen in the kernel density estimate plot below. The modified dataset had a flatter target density curve than the original density plot, which was more concentrated and peaked between pKi values of 6 and 8. The range of pKi values for the ten max phase 4 compounds collected was between 4 and 8.
Then the iblr-gn training data were fitted onto another random forest regressor model.
-
-
# n_estimators = 100 by default
-# note: if wanting to use whole dataset - switch off "bootstrap" parameter by using "False"
-rfreg_gn = RandomForestRegressor(max_depth=3, random_state=1, max_features=0.3)
-rfreg_gn.fit(X_mp4_gn, y_mp4_gn)
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Modified iblr-gn testing data were also prepared and converted into a NumPy array.
-
-
# Set up X test variable with the same molecular features
-X_mp_gn_test_df = data_gn_mp_null[['mw', 'fsp3', 'n_lipinski_hba', 'n_lipinski_hbd', 'n_rings', 'n_hetero_atoms', 'n_heavy_atoms', 'n_rotatable_bonds', 'n_radical_electrons', 'tpsa', 'qed', 'clogp', 'sas', 'n_aliphatic_carbocycles', 'n_aliphatic_heterocyles', 'n_aliphatic_rings', 'n_aromatic_carbocycles', 'n_aromatic_heterocyles', 'n_aromatic_rings', 'n_saturated_carbocycles', 'n_saturated_heterocyles', 'n_saturated_rings']]
-
-# Convert X test variables from df to arrays
-X_mp_gn_test = X_mp_gn_test_df.to_numpy()
-
-X_mp_gn_test
Using trained model for prediction on testing data
-
Predicting max phase-splitted data only.
-
-
# Predict pKi values for the compounds with "null" max phase
-# using the training model rfreg
-# Uncomment code below to print prediction result
-#print(rfreg.predict(X_mp_test))
-
-# or use:
-y_mp_test = rfreg.predict(X_mp_test)
-
-
Predicting iblr-gn data with max phase splits.
-
-
y_mp_gn_test = rfreg_gn.predict(X_mp_gn_test)
-
-
-
-
-
Scoring and metrics of trained models
-
Checking model accuracy for both training and testing datasets was recommended to take place before moving onto discovering feature importances. A scikit-learn explanation for this could be found in the section on “Permutation feature importance”. So the accuracy scores for the model were shown below.
-
-
# Training set accuracy
-print(f"Random forest regressor training accuracy: {rfreg.score(X_mp4, y_mp4):.2f}")
-
-# Testing set accuracy
-print(f"Random forest regressor testing accuracy: {rfreg.score(X_mp_test, y_mp_test):.2f}")
-
-
Random forest regressor training accuracy: 0.86
-Random forest regressor testing accuracy: 1.00
-
-
-
It looked like both the training and testing accuracies for the random forest regressor model (rfreg) were quite high, meaning that the model was able to remember the molecular features well from the training set (the tiny sample of 10 compounds), and the model was able to apply them to the testing set (which should contain about 400 or so compounds) as well, in order to make predictions on the target value of pKi. This has somewhat confirmed that the model was indeed making predictions, rather than not making any predictions at all, which meant there might be no point in finding out which features were important in the data. Therefore, we could now move onto processing the feature importances to fill in the bigger story i.e. which features were more pivotal towards influencing pKi values of approved drugs targeting acetylcholinesterase (AChE).
-
Similar model accuracy scores were also generated for the iblr-gn modified dataset, which appeared to follow a similar pattern as the max phase-splitted dataset.
-
-
# iblr-Gaussian noise & max phase splitted data
-# Training set accuracy
-print(f"Random forest regressor training accuracy: {rfreg_gn.score(X_mp4_gn, y_mp4_gn):.2f}")
-
-# Testing set accuracy
-print(f"Random forest regressor testing accuracy: {rfreg_gn.score(X_mp_gn_test, y_mp_gn_test):.2f}")
-
-
Random forest regressor training accuracy: 0.79
-Random forest regressor testing accuracy: 1.00
-
-
-
Now, setting up the y_true, which was the acutal pKi values of the testing set, and were converted into a NumPy array too.
I also found out the mean squared error (MSE) between y_true (actual max phase null compounds’ pKi values) and y_pred (predicted max phase null compounds’ pKi values). When MSE was closer to zero, the better the model was, meaning less errors were present.
-
Some references that might help with explaining MSE:
# For max phase splitted dataset only
-mean_squared_error(y_true, y_mp_test)
-
-
2.3988097789702505
-
-
-
When R2 (coefficient of determination) was closer to 1, the better the model is, with a usual range between 0 and 1 (Bruce, Bruce, and Gedeck 2020). If it was negative, then the model might not be performing as well as expected. However, there could be exceptions as other model evaluation methods should also be interpreted together with R2 (a poor R2 might not be wholly indicating it’s a poor model).
-
Some references that might help with understanding R2:
# For max phase splitted dataset only
-r2_score(y_true, y_mp_test)
-
-
-0.16228227953132635
-
-
-
Because the data was re-sampled in a iblr-gn way, the size of array would be different from the original dataset, so here I’ve specifically grabbed pKi values from the iblr-gn modified data to get the actual pKi values for the max phase null compounds.
# MSE for iblr-gn data
-mean_squared_error(y_true_gn, y_mp_gn_test)
-
-
5.7895732090189185
-
-
-
-
# R squared for iblr-gn data
-r2_score(y_true_gn, y_mp_gn_test)
-
-
-0.7425920410726885
-
-
-
Well, it appeared iblr-gn dataset might not offer much advantage than the original max phase splitted method. However, even the max phase splitted method wasn’t that great either, but it might still be interesting to find out which features were important in relation to the pKi values.
-
-
-
-
-
Feature importances
-
There were two types of feature importances available in scikit-learn, which I’ve described below. I’ve also added a Shapley additive explanations (SHAP) approach to this section as well to show different visualisation styles for feature importances on the same set of data.
-
-
-
feature_importances_ attribute from scikit-learn
-
The impurity-based feature importances (also known as Gini importance) were shown below.
-
-
# Compute feature importances on rfreg training model
-feature_imp = rfreg.feature_importances_
-
-
-
# Check what feature_imp looks like (an array)
-feature_imp
I decided to write a function to convert a NumPy array into a plot below as this was also needed in the next section.
-
-
# Function to convert array to df leading to plots
-# - for use in feature_importances_ & permutation_importance
-
-def feat_imp_plot(feat_imp_array, X_df):
-
-"""
- Function to convert feature importance array into a dataframe,
- which is then used to plot a bar graph
- to show the feature importance ranking in the random forest model for the dataset used.
-
- feat_imp_array is the array obtained from the feature_importances_ attribute,
- after having a estimator/model fitted.
-
- X_df is the dataframe for the X variable,
- where the feature column names will be used in the plot.
- """
-
-# Convert the feat_imp array into dataframe
- feat_imp_df = pd.DataFrame(feat_imp_array)
-
-# Obtain feature names via column names of dataframe
-# Rename the index as "features"
- feature = X_df.columns.rename("features")
-
-# Convert the index to dataframe
- feature_name_df = feature.to_frame(index =False)
-
-# Concatenate feature_imp_df & feature_name_df
- feature_df = pd.concat(
- [feat_imp_df, feature_name_df],
- axis=1
- ).rename(
-# Rename the column for feature importances
- columns = {0: "feature_importances"}
- ).sort_values(
-# Sort values of feature importances in descending order
-"feature_importances", ascending=False
- )
-
-# Seaborn bar plot
- sns.barplot(
- feature_df,
- x ="feature_importances",
- y ="features")
-
-
-
# Testing feat_imp_plot function
-feat_imp_plot(feature_imp, X_mp4_df)
-
-
-
-
-
An alternative way to plot was via Matplotlib directly (note: Seaborn was built based on Matplotlib, so the plots were pretty similar). The code below were probably a bit more straightforward but without axes named and the values were not sorted (only as an example but more code could be added to do this).
There were known issues with the built-in feature_importances_ attribute in scikit-learn. As quoted from scikit-learn on feature importance evaluation:
-
-
… The impurity-based feature importances computed on tree-based models suffer from two flaws that can lead to misleading conclusions. First they are computed on statistics derived from the training dataset and therefore do not necessarily inform us on which features are most important to make good predictions on held-out dataset. Secondly, they favor high cardinality features, that is features with many unique values. Permutation feature importance is an alternative to impurity-based feature importance that does not suffer from these flaws. …
-
-
So I’ve also tried the permutation_importance function (a model-agnostic method).
-
-
perm_result = permutation_importance(rfreg, X_mp_test, y_mp_test, n_repeats=10, random_state=1, n_jobs=2)
-
-# Checking data type of perm_result
-type(perm_result)
-
-
sklearn.utils._bunch.Bunch
-
-
-
It normally returns a dictionary-like objects (e.g. Bunch) with the following 3 attributes:
-
-
importances_mean (mean of feature importances)
-
importances_std (standard deviation of feature importances)
For details on these attributes, this scikit-learnlink will add a bit more explanations.
-
I decided to only use importances_mean for now.
-
-
perm_imp = perm_result.importances_mean
-
-# Confirm it produces an array
-type(perm_imp)
-
-
numpy.ndarray
-
-
-
-
# Using the function feat_imp_plot() on perm_imp result to show plot
-feat_imp_plot(perm_imp, X_mp4_df)
-
-
-
-
-
It generated a different feature importances ranking (if looking at top 6 features), although somewhat similar to the previous one.
-
-
-
-
SHAP approach
-
SHAP values (Lundberg et al. 2020), (Shapley et al. 1953) were used here to provide another way to figure out feature importances. The GitHub repository for this SHAP approach could be accessed here.
-
SHAP’s TreeExplainer() was based on Tree SHAP algorithms (Lundberg et al. 2020), and was used to show and explain feature importances within tree models. It could also be extended to boosted tree models such as LightGBM and XGBoost and also other tree models (as explained by the GitHub repository README.md and its documentation link provided). It was also a model-agnostic method, which could be quite handy.
shap_explainer = shap.TreeExplainer(rfreg)
-
-# X_test needs to be a dataframe (not numpy array)
-# otherwise feature names won't show in plot
-shap_values = shap_explainer.shap_values(X_mp_test_df)
-
-# Horizontal bar plot
-shap.summary_plot(shap_values, X_mp_test_df, plot_type ="bar")
-
-
-
-
-
Dot plot version:
-
-
shap.summary_plot(shap_values, X_mp_test_df)
-
-
-
-
-
Violin plot:
-
-
shap.summary_plot(shap_values, X_mp_test_df, plot_type ="violin")
-
-# Alternative plot option: "layered_violin"
-
-
-
-
-
-
-
-
-
Hyperparameter tuning
-
An example was shown below on tuning the number of trees (n_estimators) used in the random forest model.
-
-
# Function code adapted with thanks from ML Mastery
-# https://machinelearningmastery.com/random-forest-ensemble-in-python/
-
-# ---Evaluate a list of models with different number of trees---
-
-# Define dataset by using the same training dataset as above
-X, y = X_mp4, y_mp4
-
-# Define function to generate a list of models with different no. of trees
-def models():
-# Create empty dictionary (key, value pairs) for models
- models =dict()
-# Test different number of trees to evaluate
- no_trees = [50, 100, 250, 500, 1000]
-for n in no_trees:
- models[str(n)] = RandomForestRegressor(n_estimators=n)
-return models
-
-
-# Define function to evaluate a single model using cross-validation
-def evaluate(model, X, y):
-
-# RepeatedStratifiedKFold usually for binary or multi-class labels
-# - ref link: https://scikit-learn.org/stable/modules/generated/sklearn.model_selection.KFold.html#sklearn.model_selection.KFold
-# so using ReaptedKFold instead
- cross_val = RepeatedKFold(n_splits=10, n_repeats=15, random_state=1)
-# Run evaluation process & collect cv scores
-# Since estimator/model was based on DecisionTreeRegressor,
-# using neg_mean_squared_error metric
-# n_jobs = -1 meaning using all processors to run jobs in parallel
- scores = cross_val_score(model, X, y, scoring="neg_mean_squared_error", cv=cross_val, n_jobs=-1)
-return scores
-
-
-# Evaluate results
-# Run models with different RepeatedKFold & different no. of tress
-# with results shown as diff. trees with calculated mean cv scores & std
-
-# Obtain diff. models with diff. trees via models function
-models = models()
-
-# Create empty lists for results & names
-results, names =list(), list()
-
-# Create a for loop to iterate through the list of diff. models
-for name, model in models.items():
-# Run the cross validation scores via evaluate function
- scores = evaluate(model, X, y)
-# Collect results
- results.append(scores)
-# Collect names (different no. of trees)
- names.append(name)
-# Show the average mean squared errors and corresponding standard deviations
-# for each model with diff. no. of trees
-print((name, mean(scores), std(scores)))
-
-
('50', -1.6470594650953017, 1.6444082604560304)
-
-
-
('100', -1.6995136024743887, 1.6797340671624852)
-
-
-
('250', -1.6716290617106646, 1.6236808789148038)
-
-
-
('500', -1.645981936868625, 1.615445700037851)
-
-
-
('1000', -1.6532678610618743, 1.604259597928101)
-
-
-
The negated version of the mean squared error (neg_mean_squared_error) was due to how the scoring parameter source code was written in scikit-learn. It was written this way to take into account of both scoring and loss functions (links provided below for further explanations). All scoring metrics could be accessed here for scikit-learn.
-
Reference links to help with understanding neg_mean_squared_error:
Also, the random forest algorithm was stochastic in nature, meaning that every time hyperparameter tuning took place, it would generate different scores due to random bootstrap sampling. The best approach to evaluate model performance during the cross-validation process was to use the average outcome from several runs of cross-validations, then fit the hyperparameters on a final model, or getting several final models ready and then obtaining the average from these models instead.
-
Below was a version of boxplot plotted using Matplotlib showing the differences in the distributions of the cross validation scores and mean squared errors between different number of trees.
To plot this in Seaborn, I had to prepare the data slightly differently to achieve a different version of the boxplot. Matplotlib was a bit more straightforward to use without these steps.
-
I also used natural sort to sort numerical values (GitHub repository). Otherwise, if using sort_values() only, it would only sort the numbers in lexicographical order (i.e. by first digit only), which was not able to show the tree numbers in ascending order.
-
-
# Combine results & names lists into dataframe
-cv_results = pd.DataFrame(results, index = [names])
-
-
-
# Reset index and rename the number of trees column
-cv_results = cv_results.reset_index().rename(columns={"level_0": "Number_of_trees"})
-
-
-
# Melt the dataframe by number of trees column
-cv_results = cv_results.melt(id_vars="Number_of_trees")
-
-
-
# Sort by the number of trees column
-cv_results = cv_results.sort_values(
- by="Number_of_trees",
- key=lambda x: np.argsort(index_natsorted(cv_results["Number_of_trees"]))
-)
The Seaborn boxplot shown should be very similar to the Matplotlib one.
-
Other hyperparameters that could be tuned included:
-
-
tree depths (max_depth)
-
number of samples (max_samples)
-
number of features (max_features) - I didn’t use RDKit to generate molecular features for this post (Datamol version was used instead) which would provide around 209 at least (trying to keep the post at a readable length), but I think this might be a better option when doing cross-validations in model evaluations
-
number of nodes (max_leaf_nodes)
-
-
I’ve decided not to code for these other hyperparameters in the cross-validation step due to length of post (the function code used in cross-validation above could be further adapted to cater for other hyperparameters mentioned here), but they should be looked into if doing full-scale and comprehensive ML using the ensemble random forest algorithm.
-
-
-
-
Final words
-
Random forest was known to be a black-box ML algorithm (Bruce, Bruce, and Gedeck 2020), which was completely different from the white-box ML style revealed in decision tree graphs. Feature importances was therefore crucial to shed some lights and remove some layers of the black-box nature in random forest by showing which features were contributing towards model accuracy by ranking features used to train the model. Cross-validation was also vital to avoid over-fitting (which was more applicable to depth of trees), although in some other cases (e.g. number of trees), it was mentioned that it was unlikely the model would be overfitted. Other options available in scikit-learn ensemble methods that I didn’t get time to try were using voting classifier/regressor and stacking models to reduce biases in models, which might be very useful in other cases.
-
Few things I’ve thought of that I could try to improve what I did here was that I should really look for a different set of testing data, rather than using the max phase splits, which was not that ideal. However, as a lot of us are aware, good drug discovery data are hard to come by (a long-standing and complicated problem), I probably need some luck while looking for a different set of drug discovery data later. Another approach that I could try was that I could use RandomForestClassifier() instead on max phase prediction of these small molecules, rather than making pKi value predictions. This might involve re-labelling the max phases for these compounds into a binary or class labels, then I could use the imbalance-learn package to try and alleviate the problem with imbalanced datasets. Nevertheless, I had some fun working on this post and learnt a lot while doing it, and I hope some of the readers might find this post helpful or informative at least.
-
-
-
-
Acknowledgement
-
I’d like to thank all the authors, developers and contributors who worked towards all of the open-source packages or libraries used in this post. I’d also like to thank all of the other senior cheminformatics and ML practitioners who were sharing their work and knowledge online.
-
-
-
-
-
-
References
-
-Breiman, Leo. 1998. “Arcing Classifier (with Discussion and a Rejoinder by the Author).”The Annals of Statistics 26 (3). https://doi.org/10.1214/aos/1024691079.
-
-Chawla, N. V., K. W. Bowyer, L. O. Hall, and W. P. Kegelmeyer. 2002. “SMOTE: Synthetic Minority over-Sampling Technique.”Journal of Artificial Intelligence Research 16 (June): 321–57. https://doi.org/10.1613/jair.953.
-
-
-Kunz, Nicholas. 2020. SMOGN: Synthetic Minority over-Sampling Technique for Regression with Gaussian Noise (version v0.1.2). PyPI. https://pypi.org/project/smogn/.
-
-
-Lundberg, Scott M., Gabriel Erion, Hugh Chen, Alex DeGrave, Jordan M. Prutkin, Bala Nair, Ronit Katz, Jonathan Himmelfarb, Nisha Bansal, and Su-In Lee. 2020. “From Local Explanations to Global Understanding with Explainable AI for Trees.”Nature Machine Intelligence 2 (1): 2522–5839.
-
-
-Shapley, Lloyd S et al. 1953. “A Value for n-Person Games.”
-
-
-Torgo, Luís, Rita P. Ribeiro, Bernhard Pfahringer, and Paula Branco. 2013. “SMOTE for Regression.” In, 378–89. Springer Berlin Heidelberg. https://doi.org/10.1007/978-3-642-40669-0_33.
-
-
-Wu, Wenglei, Nicholas Kunz, and Paula Branco. 2022. “ImbalancedLearningRegression-a Python Package to Tackle the Imbalanced Regression Problem.” In Joint European Conference on Machine Learning and Knowledge Discovery in Databases, 645–48. Springer.
-
-
]]>
- Machine learning projects
- Tree models
- Pandas
- Scikit-learn
- ChEMBL database
- Python
- https://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/1_random_forest.html
- Tue, 21 Nov 2023 11:00:00 GMT
-
-
- Decision tree
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/1_data_col_prep.html
-
-
Series overview
-
-
Post 1 (this post) - data collection from ChEMBL database using web resource client in Python, with initial data preprocessing
-
Post 2 - more data preprocessing and transformation to reach the final dataset prior to model building
-
Post 3 - estimating experimental errors and building decision tree model using scikit-learn
-
-
-
-
-
Introduction
-
I’ve now come to a stage to do some more machine learning (ML) work after reading a few peer-reviewed papers about ML and drug discovery. It seemed that traditional ML methods were still indispensible performance-wise, and when used in combination with deep learning neural networks, they tend to increase prediction accuracy more. I also haven’t ventured into the practicality and usefulness of large language models in drug discovery yet, but I’m aware work in this area has been started. However, comments from experienced seniors did mention that they are still very much novel and therefore may not be as useful yet. Although by the speed of how things evolve in the so-called “AI” field, this possibly may change very soon. Also from what I can imagine, molecular representations in texts or strings are not quite the same as natural human language texts, since there are a lot of other chemistry-specific features to consider, e.g. chiralities, aromaticities and so on. Because of this, I’m sticking with learning to walk first by trying to cover conventional ML methods in a more thorough way, before trying to run in the deep learning zone.
-
So this leads to this series of posts (3 in total) about decision tree. Previously, I’ve only lightly touched on a commonly used classifier algorithm, logistic regression, as the first series in the ML realm. Reflecting back, I think I could’ve done a more thorough job during the data preparation stage. So this would be attempted this time. The data preparation used here was carried out with strong reference to the materials and methods section in this paper (Tilborg, Alenicheva, and Grisoni 2022), which was one of the papers I’ve read. There are probably other useful methods out there, but this paper made sense to me, so I’ve adopted a few of their ways of doing things during data preprocessing.
-
-
-
-
Data retrieval
-
This time I decided to try something new which was to use the ChEMBL web resource client to collect data (i.e. not by direct file downloads from ChEMBL website, although other useful way could be through SQL queries, which is also on my list to try later). I found this great online resource about fetching data this way from the TeachOpenCADD talktorial on compound data acquisition. The data retrieval workflow used below was mainly adapted from this talktorial with a few changes to suit the selected dataset and ML model.
-
The web resource client was supported by the ChEMBL group and was based on a Django QuerySet interface. Their GitHub repository might explain a bit more about it, particularly the Jupyter notebook link provided in the repository would help a lot regarding how to write code to search for specific data.
-
To do this, a few libraries needed to be loaded first.
-
-
# Import libraries
-# Fetch data through ChEMBL web resource client
-from chembl_webresource_client.new_client import new_client
-
-# Dataframe library
-import pandas as pd
-
-# Progress bar
-from tqdm import tqdm
-
-
To see what types of data were provided by ChEMBL web resource client, run the following code and refer to ChEMBL documentations to find out what data were embedded inside different data categories. Sometimes, it might not be that straight forward and some digging would be required (I went back to this step below to find the “data_validity_comment” when I was trying to do some compound sanitisations actually).
-
-
-
-
-
-
-Note
-
-
-
-
The link provided above also talked about other useful techniques for data checks in the ChEMBL database - a very important step to do during data preprocessing, which was also something I was trying to cover and achieve as much as possible in this post.
-
-
-
-
available_resources = [resource for resource indir(new_client) ifnot resource.startswith('_')]
-print(available_resources)
Resource objects were created to enable API access as suggested by the talktorial.
-
-
# for targets (proteins)
-targets_api = new_client.target
-
-# for bioactivities
-bioact_api = new_client.activity
-
-# for assays
-assay_api = new_client.assay
-
-# for compounds
-cpd_api = new_client.molecule
-
-
Checked object type for one of these API objects (e.g. bioactivity API object).
-
-
type(bioact_api)
-
-
chembl_webresource_client.query_set.QuerySet
-
-
-
-
-
-
Fetching target data
-
A protein target e.g. acetylcholinesterase was randomly chosen by using UniProt to look up the protein UniProt ID.
-
-
# Specify Uniprot ID for acetylcholinesterase
-uniprot_id ="P22303"
-
-# Get info from ChEMBL about this protein target,
-# with selected features only
-targets = targets_api.get(target_components__accession = uniprot_id).only(
-"target_chembl_id",
-"organism",
-"pref_name",
-"target_type"
-)
-
-
The query results were stored in a “targets” object, which was a QuerySet with lazy data evaluation only, meaning it would only react when there was a request for the data. Therefore, to see the results, the “targets” object was then read through Pandas DataFrame.
-
-
# Read "targets" with Pandas
-targets = pd.DataFrame.from_records(targets)
-targets
-
-
-
-
-
-
-
-
-
organism
-
pref_name
-
target_chembl_id
-
target_type
-
-
-
-
-
0
-
Homo sapiens
-
Acetylcholinesterase
-
CHEMBL220
-
SINGLE PROTEIN
-
-
-
1
-
Homo sapiens
-
Acetylcholinesterase
-
CHEMBL220
-
SINGLE PROTEIN
-
-
-
2
-
Homo sapiens
-
Cholinesterases; ACHE & BCHE
-
CHEMBL2095233
-
SELECTIVITY GROUP
-
-
-
-
-
-
-
Selected the first protein target from this dataframe.
-
-
# Save the first protein in the dataframe
-select_target = targets.iloc[0]
-select_target
-
-
organism Homo sapiens
-pref_name Acetylcholinesterase
-target_chembl_id CHEMBL220
-target_type SINGLE PROTEIN
-Name: 0, dtype: object
-
-
-
Then saved the selected ChEMBL ID for the first protein (to be used later).
Checked total rows and columns in the bioactivities dataframe.
-
-
bioact_df.shape
-
-
(706, 15)
-
-
-
-
-
Preprocess bioactivity data
-
When I reached the second half of data preprocessing, an alarm bell went off regarding using half maximal inhibitory concentration (IC50) values in ChEMBL. I remembered reading recent blog posts by Greg Landrum about using IC50 and inhibition constant (Ki) values from ChEMBL. A useful open-access paper (Kalliokoski et al. 2013) from 2013 also looked into this issue about using mixed IC50 data in ChEMBL, and provided a thorough overview about how to deal with situations like this. There was also another paper (Kramer et al. 2012) on mixed Ki data from the same author group in 2012 that touched on similar issues.
-
To summarise both the paper about IC50 and blog posts mentioned above:
-
-
it would be the best to check the details of assays used to test the compounds to ensure they were aligned and not extremely heterogeneous, since IC50 values were very assay-specific, and knowing that these values were extracted from different papers from different labs all over the world, mixing them without knowing was definitely not a good idea
-
the slightly better news was that it was more likely okay to combine Ki values for the same protein target from ChEMBL as they were found to be adding less noise to the data (however ideally similar data caution should also apply)
-
it was also possible to mix Ki values with IC50 values, but the data would need to be corrected via using a conversion factor of 2.0 to convert Ki values to IC50 values (note: I also wondered if this needed to be re-looked again since this paper was published 10 years ago…)
-
-
Because of this, I decided to stick with Ki values only for now before adding more complexities as I wasn’t entirely confident about mixing IC50 values with Ki values yet. Firstly, I checked for all types of units being used in bioact_df. There were numerous different units and formats, which meant they would need to be converted to nanomolar (nM) units first.
It looked like there were duplicates of columns on units and values, so the “units” and “value” columns were removed and “standard_units” and “standard_value” columns were kept instead. Also, “type” column was dropped as there were already a “standard_type” column.
-
-
-
-
-
-
-Note
-
-
-
-
Differences between “type” and “standard_type” columns were mentioned by this ChEMBL blog post.
Then the next step was taking care of any missing entries by removing them in the first place. I excluded “data_validity_comment” column here as this was required to check if there were any unusual activity data e.g. excessively low or high Ki values. A lot of the compounds in this column probably had empty cells or “None”, which ensured that there were no particular alarm bells to the extracted bioactivity data.
-
-
bioact_df.dropna(subset = ["activity_id", "assay_chembl_id", "assay_description", "assay_type", "molecule_chembl_id", "relation", "standard_type", "standard_units", "standard_value", "target_chembl_id", "target_organism"], axis =0, how ="any", inplace =True)
-# Check number of rows and columns again (in this case, there appeared to be no change for rows)
-bioact_df.shape
-
-
(706, 12)
-
-
-
Since all unique units inside the “units” and “values” columns were checked previously, I’d done the same for the “standard_units” column to see the ones recorded in it.
One final check on the number of columns and rows after preprocessing the bioactivity dataframe.
-
-
bioact_df.shape
-
-
(540, 12)
-
-
-
There were a total of 12 columns with 540 rows of data left in the bioactivity dataframe.
-
-
-
-
-
Fetching assay data
-
The assay data was added after I went through the rest of the data preprocessing and also after remembering to check on the confidence scores for assays used in the final data collected (to somewhat assess assay-to-target relationships). This link from ChEMBL explained what the confidence score meant.
-
-
assays = assay_api.filter(
-# Use the previously saved target ChEMBL ID
- target_chembl_id = chembl_id,
-# Binding assays only as before
- assay_type ="B"
-).only(
-"assay_chembl_id",
-"confidence_score"
-)
-
-
Placing the fetched assay data into a Pandas DataFrame.
It looked like the lowest confidence score for this particular protein target in binding assays was at 8, with others sitting at 9 (the highest). There were 452 assays with confidence score of 8.
-
-
# Some had score of 8 - find out which ones
-assays_df[assays_df["confidence_score"] ==8]
-
-
-
-
-
-
-
-
-
assay_chembl_id
-
confidence_score
-
-
-
-
-
0
-
CHEMBL634034
-
8
-
-
-
1
-
CHEMBL642512
-
8
-
-
-
2
-
CHEMBL642513
-
8
-
-
-
3
-
CHEMBL642514
-
8
-
-
-
4
-
CHEMBL642515
-
8
-
-
-
...
-
...
-
...
-
-
-
1141
-
CHEMBL3887379
-
8
-
-
-
1142
-
CHEMBL3887855
-
8
-
-
-
1143
-
CHEMBL3887947
-
8
-
-
-
1144
-
CHEMBL3888161
-
8
-
-
-
1874
-
CHEMBL5058677
-
8
-
-
-
-
452 rows × 2 columns
-
-
-
-
-
-
-
Combining bioactivity & assay data
-
The key was to combine the bioactivity and assay data along the “assay_chembl_id” column.
I actually came back to this step to relax the confidence score limit to include all the 8s as well as the 9s (otherwise previously I tried only using assays with score of 9), so that donepezil and galantamine could be included in the dataset as well (the purpose of this would be clearer in post 3 when building the model).
-
-
-
-
Fetching compound data
-
While having identified the protein target, obtained the bioactivity data, and also the assay data, this next step was to fetch the compound data. This could be done by having the ChEMBL IDs available in the bioactivity dataset.
Here, the same step was applied where the compound QuerySet object was converted into a Pandas dataframe. However, the compound data extracted here might take longer than the bioactivity one. One way to monitor progress was through using tqdm package.
Removing any missing entries in the compound data (excluding the “max_phase” column as it was needed during the model training/testing part in post 3 - note: “None” entries meant they were preclinical molecules so not assigned with a max phase yet).
-
-
cpds_df.dropna(subset = ["molecule_chembl_id", "molecule_structures"], axis =0, how ="any", inplace =True)
-
-# Check columns & rows in df
-cpds_df.shape
Ideally, only the compounds with canonical SMILES would be kept. Checking for the types of molecular representations used in the “molecule_structures” column of the compound dataset.
-
-
# Randomly choosing the 2nd entry as example
-cpds_df.iloc[1].molecule_structures.keys()
There were 4 types: “canonical_smiles”, “molfile”, “standard_inchi” and “standard_inchi_key”.
-
-
# Create an empty list to store the canonical smiles
-can_smiles = []
-
-# Create a for loop to loop over each row of data,
-# searching for only canonical_smiles to append to the created list
-for i, cpd in cpds_df.iterrows():
-try:
- can_smiles.append(cpd["molecule_structures"]["canonical_smiles"])
-exceptKeyError:
- can_smiles.append(None)
-
-# Create a new df column with name as "smiles",
-# which will store all the canonical smiles collected from the list above
-cpds_df["smiles"] = can_smiles
-
-
Check the compound dataframe quickly to see if a new column for SMILES has been created.
-
-
cpds_df.head(3)
-
-
-
-
-
-
-
-
-
max_phase
-
molecule_chembl_id
-
molecule_structures
-
smiles
-
-
-
-
-
0
-
None
-
CHEMBL28
-
{'canonical_smiles': 'O=c1cc(-c2ccc(O)cc2)oc2c...
-
O=c1cc(-c2ccc(O)cc2)oc2cc(O)cc(O)c12
-
-
-
1
-
3.0
-
CHEMBL50
-
{'canonical_smiles': 'O=c1c(O)c(-c2ccc(O)c(O)c...
-
O=c1c(O)c(-c2ccc(O)c(O)c2)oc2cc(O)cc(O)c12
-
-
-
2
-
None
-
CHEMBL8320
-
{'canonical_smiles': 'O=C1C=CC(=O)C=C1', 'molf...
-
O=C1C=CC(=O)C=C1
-
-
-
-
-
-
-
Once confirmed, the old “molecule_structures” column was then removed.
Clearly, the column that existed in both dataframes was the “molecule_chembl_id” column.
-
The next step was to combine or merge both datasets.
-
-
# Create a final dataframe that will contain both bioactivity and compound data
-dtree_df = pd.merge(
- bioact_assay_df[["molecule_chembl_id","Ki", "units", "data_validity_comment"]],
- cpds_df,
- on ="molecule_chembl_id",
-)
-
-dtree_df.head(3)
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
Ki
-
units
-
data_validity_comment
-
max_phase
-
smiles
-
-
-
-
-
0
-
CHEMBL11805
-
0.104
-
nM
-
Potential transcription error
-
None
-
COc1ccccc1CN(C)CCCCCC(=O)N(C)CCCCCCCCN(C)C(=O)...
-
-
-
1
-
CHEMBL60745
-
1.630
-
nM
-
None
-
None
-
CC[N+](C)(C)c1cccc(O)c1.[Br-]
-
-
-
2
-
CHEMBL208599
-
0.026
-
nM
-
None
-
None
-
CCC1=CC2Cc3nc4cc(Cl)ccc4c(N)c3[C@@H](C1)C2
-
-
-
-
-
-
-
Shape of the final dataframe was checked.
-
-
print(dtree_df.shape)
-
-
(540, 6)
-
-
-
Saving a copy of the merged dataframe for now to avoid re-running the previous code repeatedly, and also to be ready for second-half of the data preprocessing work, which will be in post 2.
-
-
dtree_df.to_csv("ache_chembl.csv")
-
-
-
-
-
-
-
References
-
-Kalliokoski, Tuomo, Christian Kramer, Anna Vulpetti, and Peter Gedeck. 2013. “Comparability of Mixed IC50 Data A Statistical Analysis.” Edited by Andrea Cavalli. PLoS ONE 8 (4): e61007. https://doi.org/10.1371/journal.pone.0061007.
-
-
-Kramer, Christian, Tuomo Kalliokoski, Peter Gedeck, and Anna Vulpetti. 2012. “The Experimental Uncertainty of Heterogeneous Public Ki Data.”Journal of Medicinal Chemistry 55 (11): 5165–73. https://doi.org/10.1021/jm300131x.
-
-
-Tilborg, Derek van, Alisa Alenicheva, and Francesca Grisoni. 2022. “Exposing the Limitations of Molecular Machine Learning with Activity Cliffs.”Journal of Chemical Information and Modeling 62 (23): 5938–51. https://doi.org/10.1021/acs.jcim.2c01073.
-
-
]]>
- Machine learning projects
- Tree models
- Data preprocessing
- Pandas
- ChEMBL database
- Python
- https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/1_data_col_prep.html
- Mon, 18 Sep 2023 12:00:00 GMT
-
-
- Decision tree
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/2_data_prep_tran.html
-
-
Data source
-
The data used in this post 2 for data preprocessing was extracted from ChEMBL database by using ChEMBL web resource client in Python. The details of all the steps taken to reach the final .csv file could be seen in post 1.
-
-
-
-
Checklist for preprocessing ChEMBL compound data
-
Below was a checklist summary for post 1 and post 2 (current post), and was highly inspired by this journal paper (Tilborg, Alenicheva, and Grisoni 2022) and also ChEMBL’s FAQ on “Assay and Activity Questions”.
-
Note: not an exhaustive list, only a suggestion from my experience working on this series, may need to tailor to different scenarios
-
For molecular data containing chemical compounds, check for:
-
-
duplicates
-
missing values
-
salts or mixture
-
-
Check the consistency of structural annotations:
-
-
molecular validity
-
molecular sanity
-
charge standardisation
-
stereochemistry
-
-
Check the reliability of reported experimental values (e.g. activity values like IC50, Ki, EC50 etc.):
-
-
annotated validity (data_validity_comment)
-
presence of outliers
-
confidence score (assays)
-
standard deviation of multiple entries (if applicable)
-
-
-
-
-
Import libraries
-
-
# Import all libraries used
-import pandas as pd
-import math
-from rdkit.Chem import Descriptors
-import datamol as dm
-# tqdm library used in datamol's batch descriptor code
-from tqdm import tqdm
-import mols2grid
-
-
-
-
-
Re-import saved data
-
Re-imported the partly preprocessed data from the earlier post.
There was an extra index column (named “Unnamed: 0”) here, which was likely inherited from how the .csv file was saved with the index already in place from part 1, so this column was dropped for now.
From the above quick statistical summary and also the code below to find the minimum Ki value, it confirmed there were no zero Ki values recorded.
-
-
dtree_df["Ki"].min()
-
-
0.0017
-
-
-
Now the part about converting the Ki values to pKi values, which were the negative logs of Ki in molar units (a PubChem example might help to explain it a little). The key to understand pKi here was to treat pKi similarly to how we normally understand pH for our acids and bases. The formula to convert Ki to pKi for nanomolar (nM) units was:
Applying the calc_pKi function to convert all rows of the compound dataset for the “Ki” column.
-
-
# Create a new column for pKi
-# Apply calc_pKi function to data in Ki column
-dtree_df["pKi"] = dtree_df.apply(lambda x: calc_pKi(x.Ki), axis =1)
-
-
The dataframe would now look like this, with a new pKi column (scroll to the very right to see it).
-
-
dtree_df.head(3)
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
Ki
-
units
-
data_validity_comment
-
max_phase
-
smiles
-
pKi
-
-
-
-
-
0
-
CHEMBL11805
-
0.104
-
nM
-
Potential transcription error
-
NaN
-
COc1ccccc1CN(C)CCCCCC(=O)N(C)CCCCCCCCN(C)C(=O)...
-
9.982967
-
-
-
1
-
CHEMBL60745
-
1.630
-
nM
-
NaN
-
NaN
-
CC[N+](C)(C)c1cccc(O)c1.[Br-]
-
8.787812
-
-
-
2
-
CHEMBL208599
-
0.026
-
nM
-
NaN
-
NaN
-
CCC1=CC2Cc3nc4cc(Cl)ccc4c(N)c3[C@@H](C1)C2
-
10.585027
-
-
-
-
-
-
-
-
-
-
Plan other data preprocessing steps
-
For a decision tree model, a few more molecular descriptors were most likely needed rather than only Ki or pKi and SMILES, since I’ve now arrived at the step of planning other preprocessing steps. One way to do this could be through computations based on canonical SMILES of compounds by using RDKit, which would give the RDKit 2D descriptors. In this single tree model, I decided to stick with only RDKit 2D descriptors for now, before adding on fingerprints (as a side note: I have very lightly touched on generating fingerprints in this earlier post - “Molecular similarities in selected COVID-19 antivirals” in the subsection on “Fingerprint generator”).
-
At this stage, a compound sanitisation step should also be applied to the compound column before starting any calculations to rule out compounds with questionable chemical validities. RDKit or Datamol (a Python wrapper library built based on RDKit) was also capable of doing this.
-
I’ve added a quick step here to convert the data types of “smiles” and “data_validity_comment” columns to string (in case of running into problems later).
There were 3 different types of data validity comments here, which were “Potential transcription error”, “NaN” and “Outside typical range”. So, this meant compounds with comments as “Potential transcription error” and “Outside typical range” should be addressed first.
-
-
# Find out number of compounds with "outside typical range" as data validity comment
-dtree_df_err = dtree_df[dtree_df["data_validity_comment"] =="Outside typical range"]
-print(dtree_df_err.shape)
-dtree_df_err.head()
-
-
(58, 7)
-
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
Ki
-
units
-
data_validity_comment
-
max_phase
-
smiles
-
pKi
-
-
-
-
-
111
-
CHEMBL225198
-
0.0090
-
nM
-
Outside typical range
-
NaN
-
O=C(CCc1c[nH]c2ccccc12)NCCCCCCCNc1c2c(nc3cc(Cl...
-
11.045757
-
-
-
114
-
CHEMBL225021
-
0.0017
-
nM
-
Outside typical range
-
NaN
-
O=C(CCCc1c[nH]c2ccccc12)NCCCCCNc1c2c(nc3cc(Cl)...
-
11.769551
-
-
-
118
-
CHEMBL402976
-
313700.0000
-
nM
-
Outside typical range
-
NaN
-
CN(C)CCOC(=O)Nc1ccncc1
-
3.503485
-
-
-
119
-
CHEMBL537454
-
140200.0000
-
nM
-
Outside typical range
-
NaN
-
CN(C)CCOC(=O)Nc1cc(Cl)nc(Cl)c1.Cl
-
3.853252
-
-
-
120
-
CHEMBL3216883
-
316400.0000
-
nM
-
Outside typical range
-
NaN
-
CN(C)CCOC(=O)Nc1ccncc1Br.Cl.Cl
-
3.499764
-
-
-
-
-
-
-
There were a total of 58 compounds with Ki outside typical range.
With the other comment for potential transciption error, there seemed to be only one compound here.
-
These compounds with questionable Ki values were removed, as they could be potential sources of errors in ML models later on (error trickling effect). One of the ways to filter out data was to fill the empty cells within the “data_validity_comment” column first, so those ones to be kept could be selected.
-
-
# Fill "NaN" entries with an actual name e.g. none
-dtree_df["data_validity_comment"].fillna("none", inplace=True)
-dtree_df.head()
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
Ki
-
units
-
data_validity_comment
-
max_phase
-
smiles
-
pKi
-
-
-
-
-
0
-
CHEMBL11805
-
0.104
-
nM
-
Potential transcription error
-
NaN
-
COc1ccccc1CN(C)CCCCCC(=O)N(C)CCCCCCCCN(C)C(=O)...
-
9.982967
-
-
-
1
-
CHEMBL60745
-
1.630
-
nM
-
none
-
NaN
-
CC[N+](C)(C)c1cccc(O)c1.[Br-]
-
8.787812
-
-
-
2
-
CHEMBL208599
-
0.026
-
nM
-
none
-
NaN
-
CCC1=CC2Cc3nc4cc(Cl)ccc4c(N)c3[C@@H](C1)C2
-
10.585027
-
-
-
3
-
CHEMBL95
-
151.000
-
nM
-
none
-
4.0
-
Nc1c2c(nc3ccccc13)CCCC2
-
6.821023
-
-
-
4
-
CHEMBL173309
-
12.200
-
nM
-
none
-
NaN
-
CCN(CCCCCC(=O)N(C)CCCCCCCCN(C)C(=O)CCCCCN(CC)C...
-
7.913640
-
-
-
-
-
-
-
Filtered out only the compounds with nil data validity comments.
This preprocessing molecules tutorial and reference links provided by Datamol were very informative, and the preprocess function code by Datamol was used below. Each step of fix_mol(), sanitize_mol() and standardize_mol() was explained in this tutorial. I think the key was to select preprocessing options required to fit the purpose of the ML models, and the more experiences in doing this, the more likely it will help with the preprocessing step.
-
-
# _preprocess function to sanitise compounds - adapted from datamol.io
-
-smiles_column ="smiles"
-
-dm.disable_rdkit_log()
-
-def _preprocess(row):
-# Convert each compound to a RDKit molecule in the smiles column
- mol = dm.to_mol(row[smiles_column], ordered=True)
-# Fix common errors in the molecules
- mol = dm.fix_mol(mol)
-# Sanitise the molecules
- mol = dm.sanitize_mol(mol, sanifix=True, charge_neutral=False)
-# Standardise the molecules
- mol = dm.standardize_mol(
- mol,
-# Switch on to disconnect metal ions
- disconnect_metals=True,
- normalize=True,
- reionize=True,
-# Switch on "uncharge" to neutralise charges
- uncharge=True,
-# Taking care of stereochemistries of compounds
- stereo=True,
- )
-
-# Added a new column below for RDKit molecules
- row["rdkit_mol"] = dm.to_mol(mol)
- row["standard_smiles"] = dm.standardize_smiles(dm.to_smiles(mol))
- row["selfies"] = dm.to_selfies(mol)
- row["inchi"] = dm.to_inchi(mol)
- row["inchikey"] = dm.to_inchikey(mol)
-return row
-
-
Then the compound sanitisation function was applied to the dtree_df.
In this case, I tried using the preprocessing function without adding parallelisation, the whole process wasn’t very long (since I had a small dataset), and was done within a minute or so.
-
Also, as a sanity check on the sanitised compounds in dtree_san_df, I just wanted to see if I could display all compounds in this dataframe as 2D images. I also had a look through each page just to see if there were any odd bonds or anything strange in general.
-
-
# Create a list to store all cpds in dtree_san_df
-mol_list = dtree_san_df["rdkit_mol"]
-# Convert to list
-mol_list =list(mol_list)
-# Check data type
-type(mol_list)
-# Show 2D compound structures in grids
-mols2grid.display(mol_list)
-
-
-
-
-
-
-
-
-
-
-
Detect outliers
-
Plotting a histogram to see the distribution of pKi values first.
I read a bit about Dixon’s Q test and realised that there were a few required assumptions prior to using this test, and the current dataset used here (dtree_san_df) might not fit the requirements, which were:
-
-
normally distributed data
-
a small sample size e.g. between 3 and 10, which was originally stated in this paper (Dean and Dixon 1951).
dtree_san_df.boxplot(column ="pKi")
-
-# the boxplot version below shows a blank background
-# rather than above version with horizontal grid lines
-#dtree_san_df.plot.box(column = "pKi")
-
-
<AxesSubplot: >
-
-
-
-
-
-
I also used Pandas’ built-in boxplot in addition to the histogram to show the possible outliers within the pKi values. Clearly, the outliers for pKi values appeared to be above 10. I also didn’t remove these outliers completely due to the dataset itself wasn’t quite in a Gaussian distribution (they might not be true outliers).
-
-
-
-
Calculate RDKit 2D molecular descriptors
-
I’ve explored a few different ways to compute molecular descriptors, essentially RDKit was used as the main library to do this (there might be other options via other programming languages, but I was only exploring RDKit-based methods via Python for now). A blog post I’ve come across on calculating RDKit 2D molecular descriptors has explained it well, it gave details about how to bundle the functions together in a class (the idea of building a small library yourself to be used in projects was quite handy). I’ve also read RDKit’s documentations and also the ones from Datamol. So rather than re-inventing the wheels of all the RDKit code, I’ve opted to use only a small chunk of RDKit code as a demonstration, then followed by Datamol’s version to compute the 2D descriptors, since there were already a few really well-explained blog posts about this. One of the examples was this useful descriptor calculation tutorial by Greg Landrum.
-
-
-
RDKit code
-
With the lastest format of the dtree_san_df, it already included a RDKit molecule column (named “rdkit_mol”), so this meant I could go ahead with the calculations. So here I used RDKit’s Descriptors.CalcMolDescriptors() to calculate the 2D descriptors - note: there might be more code variations depending on needs, this was just a small example.
-
-
# Run descriptor calculations on mol_list (created earlier)
-# and save as a new list
-mol_rdkit_ls = [Descriptors.CalcMolDescriptors(mol) for mol in mol_list]
-
-# Convert the list into a dataframe
-df_rdkit_2d = pd.DataFrame(mol_rdkit_ls)
-print(df_rdkit_2d.shape)
-df_rdkit_2d.head(3)
-
-
(481, 209)
-
-
-
-
-
-
-
-
-
-
MaxAbsEStateIndex
-
MaxEStateIndex
-
MinAbsEStateIndex
-
MinEStateIndex
-
qed
-
MolWt
-
HeavyAtomMolWt
-
ExactMolWt
-
NumValenceElectrons
-
NumRadicalElectrons
-
...
-
fr_sulfide
-
fr_sulfonamd
-
fr_sulfone
-
fr_term_acetylene
-
fr_tetrazole
-
fr_thiazole
-
fr_thiocyan
-
fr_thiophene
-
fr_unbrch_alkane
-
fr_urea
-
-
-
-
-
0
-
9.261910
-
9.261910
-
0.000000
-
0.000000
-
0.662462
-
246.148
-
230.020
-
245.041526
-
74
-
0
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
1
-
6.509708
-
6.509708
-
0.547480
-
0.547480
-
0.763869
-
298.817
-
279.665
-
298.123676
-
108
-
0
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
2
-
6.199769
-
6.199769
-
0.953981
-
0.953981
-
0.706488
-
198.269
-
184.157
-
198.115698
-
76
-
0
-
...
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
-
3 rows × 209 columns
-
-
-
-
In total, it generated 209 descriptors.
-
-
-
-
Datamol code
-
Then I tested Datamol’s code on this as shown below.
-
-
# Datamol's batch descriptor code for a list of compounds
-dtree_san_df_dm = dm.descriptors.batch_compute_many_descriptors(mol_list)
-print(dtree_san_df_dm.shape)
-dtree_san_df_dm.head(3)
-
-
(481, 22)
-
-
-
-
-
-
-
-
-
-
mw
-
fsp3
-
n_lipinski_hba
-
n_lipinski_hbd
-
n_rings
-
n_hetero_atoms
-
n_heavy_atoms
-
n_rotatable_bonds
-
n_radical_electrons
-
tpsa
-
...
-
sas
-
n_aliphatic_carbocycles
-
n_aliphatic_heterocyles
-
n_aliphatic_rings
-
n_aromatic_carbocycles
-
n_aromatic_heterocyles
-
n_aromatic_rings
-
n_saturated_carbocycles
-
n_saturated_heterocyles
-
n_saturated_rings
-
-
-
-
-
0
-
245.041526
-
0.400000
-
2
-
1
-
1
-
3
-
13
-
2
-
0
-
20.23
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
1
-
298.123676
-
0.388889
-
2
-
2
-
4
-
3
-
21
-
1
-
0
-
38.91
-
...
-
4.331775
-
2
-
0
-
2
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
2
-
198.115698
-
0.307692
-
2
-
2
-
3
-
2
-
15
-
0
-
0
-
38.91
-
...
-
2.014719
-
1
-
0
-
1
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
-
3 rows × 22 columns
-
-
-
-
There were a total of 22 molecular descriptors generated, which seemed more like what I might use for the decision tree model. The limitation with this batch descriptor code was that the molecular features were pre-selected, so if other types were needed, it would be the best to go for RDKit code or look into other Datamol descriptor code that allow users to specify features. The types of descriptors were shown below.
The trickier part for data preprocessing was actually trying to merge, join or concatenate dataframes of the preprocessed dataframe (dtree_san_df) and the dataframe from Datamol’s descriptor code (dtree_san_df_dm).
-
Initially, I tried using all of Pandas’ code of merge/join/concat() dataframes. They all failed to create the correct final combined dataframe with too many rows generated, with one run actually created more than 500 rows (maximum should be 481 rows). One of the possible reasons for this could be that some of the descriptors had zeros generated as results for some of the compounds, and when combining dataframes using Pandas code like the ones mentioned here, they might cause unexpected results (as suggested by Pandas, these code were not exactly equivalent to SQL joins). So I looked into different ways, and while there were no other common columns for both dataframes, the index column seemed to be the only one that correlated both.
-
I also found out after going back to the previous steps that when I applied the compound preprocessing function from Datamol, the index of the resultant dataframe was changed to start from 1 (rather than zero). Because of this, I tried re-setting the index of dtree_san_df first, then dropped the index column, followed by re-setting the index again to ensure it started at zero, which has worked. So now the dtree_san_df would have exactly the same index as the one for dtree_san_df_dm.
-
-
# 1st index re-set
-dtree_san_df = dtree_san_df.reset_index()
-# Drop the index column
-dtree_san_df = dtree_san_df.drop(["index"], axis =1)
-dtree_san_df.head(3)
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
Ki
-
units
-
data_validity_comment
-
max_phase
-
smiles
-
pKi
-
rdkit_mol
-
standard_smiles
-
selfies
-
inchi
-
inchikey
-
-
-
-
-
0
-
CHEMBL60745
-
1.630
-
nM
-
none
-
NaN
-
CC[N+](C)(C)c1cccc(O)c1.[Br-]
-
8.787812
-
<rdkit.Chem.rdchem.Mol object at 0x120080f90>
-
CC[N+](C)(C)c1cccc(O)c1.[Br-]
-
[C][C][N+1][Branch1][C][C][Branch1][C][C][C][=...
-
InChI=1S/C10H15NO.BrH/c1-4-11(2,3)9-6-5-7-10(1...
-
CAEPIUXAUPYIIJ-UHFFFAOYSA-N
-
-
-
1
-
CHEMBL208599
-
0.026
-
nM
-
none
-
NaN
-
CCC1=CC2Cc3nc4cc(Cl)ccc4c(N)c3[C@@H](C1)C2
-
10.585027
-
<rdkit.Chem.rdchem.Mol object at 0x120081cb0>
-
CCC1=CC2Cc3nc4cc(Cl)ccc4c(N)c3[C@@H](C1)C2
-
[C][C][C][=C][C][C][C][=N][C][=C][C][Branch1][...
-
InChI=1S/C18H19ClN2/c1-2-10-5-11-7-12(6-10)17-...
-
QTPHSDHUHXUYFE-KIYNQFGBSA-N
-
-
-
2
-
CHEMBL95
-
151.000
-
nM
-
none
-
4.0
-
Nc1c2c(nc3ccccc13)CCCC2
-
6.821023
-
<rdkit.Chem.rdchem.Mol object at 0x120081700>
-
Nc1c2c(nc3ccccc13)CCCC2
-
[N][C][=C][C][=Branch1][N][=N][C][=C][C][=C][C...
-
InChI=1S/C13H14N2/c14-13-9-5-1-3-7-11(9)15-12-...
-
YLJREFDVOIBQDA-UHFFFAOYSA-N
-
-
-
-
-
-
-
-
# 2nd index re-set
-dtree_san_df = dtree_san_df.reset_index()
-print(dtree_san_df.shape)
-dtree_san_df.head(3)
Checking final dataframe to make sure there were 481 rows (also that index_x and index_y were identical) and also there was an increased number of columns (columns combined from both dataframes). So this finally seemed to work.
-
-
print(dtree_f_df.shape)
-dtree_f_df.head(3)
-
-
(481, 27)
-
-
-
-
-
-
-
-
-
-
index_x
-
molecule_chembl_id
-
pKi
-
max_phase
-
index_y
-
mw
-
fsp3
-
n_lipinski_hba
-
n_lipinski_hbd
-
n_rings
-
...
-
sas
-
n_aliphatic_carbocycles
-
n_aliphatic_heterocyles
-
n_aliphatic_rings
-
n_aromatic_carbocycles
-
n_aromatic_heterocyles
-
n_aromatic_rings
-
n_saturated_carbocycles
-
n_saturated_heterocyles
-
n_saturated_rings
-
-
-
-
-
0
-
0
-
CHEMBL60745
-
8.787812
-
NaN
-
0
-
245.041526
-
0.400000
-
2
-
1
-
1
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
1
-
1
-
CHEMBL208599
-
10.585027
-
NaN
-
1
-
298.123676
-
0.388889
-
2
-
2
-
4
-
...
-
4.331775
-
2
-
0
-
2
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
2
-
2
-
CHEMBL95
-
6.821023
-
4.0
-
2
-
198.115698
-
0.307692
-
2
-
2
-
3
-
...
-
2.014719
-
1
-
0
-
1
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
-
3 rows × 27 columns
-
-
-
-
The two index columns (“index_x” and “index_y”) were removed, which brought out the final preprocessed dataframe.
I then saved this preprocessed dataframe as another file in my working directory, so that it could be used for estimating experimental errors and model building in the next post.
-
-
dtree_f_df.to_csv("ache_2d_chembl.csv")
-
-
-
-
-
Data preprocessing reflections
-
In general, the order of steps could be swapped in a more logical way. The subsections presented in this post bascially reflected my thought processes, as there were some back-and-forths. The whole data preprocessing step was probably still not thorough enough, but I’ve tried to cover as much as I could (hopefully I didn’t go overboard with it…). Also, it might still not be ideal to use Ki values this freely as mentioned in post 1 (noises in data issues).
-
It was mentioned in scikit-learn that for decision tree models, because of its non-parametric nature, there were not a lot of data cleaning required. However, I think that might be domain-specific, since for the purpose of drug discovery, if this step wasn’t done properly, whatever result that came out of the ML model most likely would not work and also would not reflect real-life scenarios. I was also planning on extending this series to add more trees to the model, that is, from one tree (decision tree), to multiple trees (random forests), and then hopefully move on to boosted trees (XGBoost and LightGBM). Therefore, I’d better do this data cleaning step well first to save some time later (if using the same set of data).
-
Next post will be about model building using scikit-learn and also a small part on estimating experimental errors on the dataset - this is going to be in post 3.
-
-
-
-
-
-
References
-
-Dean, R. B., and W. J. Dixon. 1951. “Simplified Statistics for Small Numbers of Observations.”Analytical Chemistry 23 (4): 636–38. https://doi.org/10.1021/ac60052a025.
-
-
-Tilborg, Derek van, Alisa Alenicheva, and Francesca Grisoni. 2022. “Exposing the Limitations of Molecular Machine Learning with Activity Cliffs.”Journal of Chemical Information and Modeling 62 (23): 5938–51. https://doi.org/10.1021/acs.jcim.2c01073.
-
-
]]>
- Machine learning projects
- Tree models
- Data preprocessing
- ChEMBL database
- Pandas
- RDKit
- Datamol
- Python
- https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/2_data_prep_tran.html
- Mon, 18 Sep 2023 12:00:00 GMT
-
-
- Decision tree
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/3_model_build.html
- Post updated on 28th April 2024 - dtreeviz code (under the “Model building” section) were updated to improve the scale of the dtreeviz tree plot
-
-
-
Data source
-
The data used here was extracted from ChEMBL database by using ChEMBL web resource client in Python. The details of all the steps taken to reach the final .csv file could be seen in these earlier posts - post 1 and post 2 (yes, it took quite a while to clean the data, so it was splitted into two posts).
-
-
The final .csv file used to train the model was named, “ache_2d_chembl.csv”
-
The earlier version without any RDKit 2D descriptors calculated was named, “ache_chembl.csv”
-
Both files should be in a GitHub repository called, “ML2-1_decision_tree” or in my blog repository, under “posts” folder (look for “16_ML2-1_Decision_tree” folder)
-
-
-
-
-
Estimate experimental errors
-
This part was about estimating the impact of experimental errors (pKi values) on the predictive machine learning (ML) models. It was also needed to estimate the maximum possible correlation that could be drawn from the dataset prepared from the previous two posts. I supposed it made more sense if this was done prior to building the ML model, so this wouldn’t be forgotten or missed, as we know that real-life is full of many imperfections.
-
This subsection was inspired by Pat Walters’ posts, which have discussed about estimating errors for experimental data with code links available in these posts:
-
-
How Good Could (Should) My Models Be? - a reference paper (Brown, Muchmore, and Hajduk 2009) was mentioned as the simulation basis for estimating the impact of experimental errors on the correlation from a predictive ML model
The pKi column was used in the code below as it contained the experimental values (calculated from measured Ki values, usually derived from countless lab experiments) collected from different scientific literatures or other sources as stated in ChEMBL. The aim was to simulate pKi values with experimental errors added to them.
-
Code used for the rest of the subsection were adapted with thanks from Pat Walters’ “maximum_correlation.ipynb” with my own added comments for further explanations
-
-
# Save exp data (pKi) as an object
-data = dtree["pKi"]
-# Save the object as a list
-data_ls = [data]
-
-# Trial 3-, 5- & 10-fold errors
-for fold in (3, 5, 10):
-# Retrieve error samples randomly from a normal distribution
-# Bewteen 0 and log10 of number-fold
-# for the length of provided data only
- error = np.random.normal(0, np.log10(fold), len(data))
- data_ls.append(error + data)
-
-# Convert data_ls to dataframe
-dtree_err = pd.DataFrame(data_ls)
-# Re-align dataframe (switch column header & index)
-dtree_err = dtree_err.transpose()
-# Rename columns
-dtree_err.columns = ["pKi", "3-fold", "5-fold", "10-fold"]
-print(dtree_err.shape)
-dtree_err.head()
-
-
(481, 4)
-
-
-
-
-
-
-
-
-
-
pKi
-
3-fold
-
5-fold
-
10-fold
-
-
-
-
-
0
-
8.787812
-
8.710912
-
9.101193
-
7.471251
-
-
-
1
-
10.585027
-
10.883334
-
10.291557
-
9.455301
-
-
-
2
-
6.821023
-
6.134753
-
6.799967
-
7.122006
-
-
-
3
-
7.913640
-
8.390146
-
7.874722
-
7.209130
-
-
-
4
-
6.698970
-
7.359148
-
7.290723
-
5.770489
-
-
-
-
-
-
-
Melting the created dtree_err so it could be plotted later (noticed there should be an increased number of rows after re-stacking the data).
-
-
# Melt the dtree_err dataframe
-# to make error values in one column (for plotting)
-dtree_err_melt = dtree_err.melt(id_vars ="pKi")
-print(dtree_err_melt.shape)
-dtree_err_melt.head()
-
-
(1443, 3)
-
-
-
-
-
-
-
-
-
-
pKi
-
variable
-
value
-
-
-
-
-
0
-
8.787812
-
3-fold
-
8.710912
-
-
-
1
-
10.585027
-
3-fold
-
10.883334
-
-
-
2
-
6.821023
-
3-fold
-
6.134753
-
-
-
3
-
7.913640
-
3-fold
-
8.390146
-
-
-
4
-
6.698970
-
3-fold
-
7.359148
-
-
-
-
-
-
-
Presenting this in regression plots.
-
Note: There was a matplotlib bug which would always show a tight_layout user warning for FacetGrid plots in seaborn (the lmplot used below). Seaborn was built based on matplotlib so unsurprisingly this occurred (this GitHub issue link might explain it). I have therefore temporarily silenced this user warning for the sake of post publication.
-
-
# To silence the tight-layout user warning
-import warnings
-warnings.filterwarnings("ignore")
-
-# variable = error-fold e.g. 3-fold
-# value = pKi value plus error
-sns.set_theme(font_scale =1.5)
-plot = sns.lmplot(
- x ="pKi",
- y ="value",
- col ="variable",
- data = dtree_err_melt,
-# alpha = mark’s opacity (low - more transparent)
-# s = mark size (increase with higher number)
- scatter_kws =dict(alpha =0.5, s =15)
- )
-title_list = ["3-fold", "5-fold", "10-fold"]
-for i inrange(0, 3):
- plot.axes[0, i].set_ylabel("pKi + error")
- plot.axes[0, i].set_title(title_list[i])
-
-
-
-
-
Simulating the impact of error on the correlation between experimental pKi and also pKi with errors (3-fold, 5-fold and 10-fold). R2 calculated using scikit-learn was introduced in the code below.
-
-
# Calculating r2 score (coefficient of determination)
-# based on 1000 trials for each fold
-# note: data = dtree["pKi"]
-
-# Create an empty list for correlation
-cor_ls = []
-for fold in [3, 5, 10]:
-# Set up 1000 trials
-for i inrange(0, 1000):
- error = np.random.normal(0, np.log10(fold), len(data))
- cor_ls.append([r2_score(data, data + error), f"{fold}-fold"])
-
-# Convert cor_ls into dataframe
-err_df = pd.DataFrame(cor_ls, columns = ["r2", "fold_error"])
-err_df.head()
This definitely helped a lot with visualising the estimated errors for the experimental Ki values curated in ChEMBL for this specific protein target (CHEMBL220, acetylcholinesterase (AChE)). The larger the error-fold, the lower the R2, and once the experimental error reached 10-fold, we could see an estimated R2 (maximum correlation) with its median sitting below 0.55, indicating a likely poor predictive ML model if it was built based on these data with the estimated 10-fold experimental errors.
-
-
-
-
Check max phase distribution
-
At this stage, I’ve planned to do model training on compounds with max phase 4 (i.e. prescription medicines), so this would somewhat be an attempt to mirror real-life scenarios for the ML prediction model.
-
Max phases were assigned to each ChEMBL-curated compound according to this ChEMBL FAQ link (under the question of “What is max phase?”). As quoted from this ChEMBL FAQ link, a max phase 4 compound means:
-
-
“Approved (4): A marketed drug e.g. AMINOPHYLLINE (CHEMBL1370561) is an FDA approved drug for treatment of asthma.”
-
-
Checking out the actual counts of each max phase group in the dataset.
There was only a very small number of compounds with max phase 4 assigned (a total count of 10, which was also unsurprising since there weren’t many AChE inhibitors used as prescription medications for dementia - some of the well-known examples were donepezil, galantamine and rivastigmine).
-
Filling in actual “null” labels for all “NaN” rows in the “max_phase” columns to help with filtering out these compounds later on.
This was just another sanity check for myself on the dtree dataframe - making sure there weren’t any “NaN” cells in it (so dropping any “NaN” again, even though I might have already done this as one of the steps during data preprocessing).
-
-
dtree.dropna()
-print(dtree.shape)
-dtree.head()
-
-
(481, 25)
-
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
pKi
-
max_phase
-
mw
-
fsp3
-
n_lipinski_hba
-
n_lipinski_hbd
-
n_rings
-
n_hetero_atoms
-
n_heavy_atoms
-
...
-
sas
-
n_aliphatic_carbocycles
-
n_aliphatic_heterocyles
-
n_aliphatic_rings
-
n_aromatic_carbocycles
-
n_aromatic_heterocyles
-
n_aromatic_rings
-
n_saturated_carbocycles
-
n_saturated_heterocyles
-
n_saturated_rings
-
-
-
-
-
0
-
CHEMBL60745
-
8.787812
-
null
-
245.041526
-
0.400000
-
2
-
1
-
1
-
3
-
13
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
1
-
CHEMBL208599
-
10.585027
-
null
-
298.123676
-
0.388889
-
2
-
2
-
4
-
3
-
21
-
...
-
4.331775
-
2
-
0
-
2
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
2
-
CHEMBL95
-
6.821023
-
4.0
-
198.115698
-
0.307692
-
2
-
2
-
3
-
2
-
15
-
...
-
2.014719
-
1
-
0
-
1
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
3
-
CHEMBL173309
-
7.913640
-
null
-
694.539707
-
0.666667
-
8
-
0
-
2
-
8
-
50
-
...
-
2.803680
-
0
-
0
-
0
-
2
-
0
-
2
-
0
-
0
-
0
-
-
-
4
-
CHEMBL1128
-
6.698970
-
4.0
-
201.092042
-
0.400000
-
2
-
1
-
1
-
3
-
13
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
-
5 rows × 25 columns
-
-
-
-
-
-
-
Model building
-
-
Training data based on max phase 4 compounds
-
So here I wanted to separate the collected data by splitting the compounds into two groups based on their assigned max phases. Compounds with max phase 4 were chosen as the training data, and the rest of the compounds with max phases of “null” would be the testing data.
-
-
# Create a df for compounds with max phase 4 only
-dtree_mp4 = dtree[dtree["max_phase"] ==4]
-dtree_mp4
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
pKi
-
max_phase
-
mw
-
fsp3
-
n_lipinski_hba
-
n_lipinski_hbd
-
n_rings
-
n_hetero_atoms
-
n_heavy_atoms
-
...
-
sas
-
n_aliphatic_carbocycles
-
n_aliphatic_heterocyles
-
n_aliphatic_rings
-
n_aromatic_carbocycles
-
n_aromatic_heterocyles
-
n_aromatic_rings
-
n_saturated_carbocycles
-
n_saturated_heterocyles
-
n_saturated_rings
-
-
-
-
-
2
-
CHEMBL95
-
6.821023
-
4.0
-
198.115698
-
0.307692
-
2
-
2
-
3
-
2
-
15
-
...
-
2.014719
-
1
-
0
-
1
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
4
-
CHEMBL1128
-
6.698970
-
4.0
-
201.092042
-
0.400000
-
2
-
1
-
1
-
3
-
13
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
6
-
CHEMBL640
-
6.000000
-
4.0
-
235.168462
-
0.461538
-
4
-
3
-
1
-
4
-
17
-
...
-
1.791687
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
9
-
CHEMBL502
-
7.688246
-
4.0
-
379.214744
-
0.458333
-
4
-
0
-
4
-
4
-
28
-
...
-
2.677222
-
1
-
1
-
2
-
2
-
0
-
2
-
0
-
1
-
1
-
-
-
131
-
CHEMBL481
-
7.296709
-
4.0
-
586.279135
-
0.515152
-
10
-
1
-
7
-
10
-
43
-
...
-
3.632560
-
0
-
4
-
4
-
1
-
2
-
3
-
0
-
2
-
2
-
-
-
133
-
CHEMBL360055
-
4.431798
-
4.0
-
510.461822
-
0.800000
-
6
-
0
-
1
-
6
-
36
-
...
-
3.257653
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
160
-
CHEMBL1025
-
5.221849
-
4.0
-
184.066459
-
1.000000
-
3
-
0
-
0
-
5
-
11
-
...
-
3.345144
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
171
-
CHEMBL659
-
6.522879
-
4.0
-
287.152144
-
0.529412
-
4
-
1
-
4
-
4
-
21
-
...
-
4.226843
-
1
-
2
-
3
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
180
-
CHEMBL1200970
-
4.607303
-
4.0
-
348.142697
-
0.368421
-
2
-
0
-
3
-
4
-
23
-
...
-
4.223591
-
0
-
1
-
1
-
2
-
0
-
2
-
0
-
0
-
0
-
-
-
195
-
CHEMBL1677
-
6.995679
-
4.0
-
234.092376
-
0.307692
-
2
-
2
-
3
-
3
-
16
-
...
-
3.218715
-
1
-
0
-
1
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
-
10 rows × 25 columns
-
-
-
-
Making sure donepezil and galantamine were in this dtree_mp4 dataframe, so the model training would be based on these medicines and also other max phase 4 AChE inhibitors.
-
The screenshots of both medicines were taken from ChEMBL website:
-
-
-
-Screenshot of donepezil (parent drug form) with its molecule ChEMBL ID
-
-
-
-
-
-Screenshot of galantamine (parent drug form) with its molecule ChEMBL ID
-
-
-
The following regex string check confirmed that these two compounds were in the dtree_mp4 dataframe - row indices 9 and 171 contained these two drugs.
The DecisionTreeRegressor() was fitted on the compounds with max phase 4 as shown below, keeping tree depth at 3 for now to avoid complicating the overall tree graph (the deeper the tree, the more branches - potentially might overfit and create noises for the model).
The following was a dtreeviz version of the decision tree, which actually included the regression plots of different molecular features e.g. clogp versus the target value of pKi. It seemed a bit more intuitive as these plots clearly showed where the threshold cut-offs would be for each feature (molecular descriptors). The GitHub repository link for dtreeviz could be accessed here.
-
-
import dtreeviz
-
-viz = dtreeviz.model(ache_tree_mp4, X_train=X_mp4, y_train=y_mp4, target_name="pKi", feature_names=list(X_mp4_df.columns))
-# Added "scale = 2" to view()
-# to make plot larger in size
-viz.view(scale =2)
-
-
-
-
-
-
-
-
Testing and predicting data based on max phase of null compounds
-
-
# Compounds with max phase as "null"
-dtree_mp_null = dtree[dtree["max_phase"] =="null"]
-print(dtree_mp_null.shape)
-dtree_mp_null.head()
-
-
(466, 25)
-
-
-
-
-
-
-
-
-
-
molecule_chembl_id
-
pKi
-
max_phase
-
mw
-
fsp3
-
n_lipinski_hba
-
n_lipinski_hbd
-
n_rings
-
n_hetero_atoms
-
n_heavy_atoms
-
...
-
sas
-
n_aliphatic_carbocycles
-
n_aliphatic_heterocyles
-
n_aliphatic_rings
-
n_aromatic_carbocycles
-
n_aromatic_heterocyles
-
n_aromatic_rings
-
n_saturated_carbocycles
-
n_saturated_heterocyles
-
n_saturated_rings
-
-
-
-
-
0
-
CHEMBL60745
-
8.787812
-
null
-
245.041526
-
0.400000
-
2
-
1
-
1
-
3
-
13
-
...
-
3.185866
-
0
-
0
-
0
-
1
-
0
-
1
-
0
-
0
-
0
-
-
-
1
-
CHEMBL208599
-
10.585027
-
null
-
298.123676
-
0.388889
-
2
-
2
-
4
-
3
-
21
-
...
-
4.331775
-
2
-
0
-
2
-
1
-
1
-
2
-
0
-
0
-
0
-
-
-
3
-
CHEMBL173309
-
7.913640
-
null
-
694.539707
-
0.666667
-
8
-
0
-
2
-
8
-
50
-
...
-
2.803680
-
0
-
0
-
0
-
2
-
0
-
2
-
0
-
0
-
0
-
-
-
5
-
CHEMBL102226
-
4.698970
-
null
-
297.152928
-
0.923077
-
3
-
0
-
0
-
5
-
18
-
...
-
2.965170
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
7
-
CHEMBL103873
-
5.698970
-
null
-
269.121628
-
0.909091
-
3
-
0
-
0
-
5
-
16
-
...
-
3.097106
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
0
-
-
-
-
5 rows × 25 columns
-
-
-
-
There were 466 compounds with max phase as “null”, meaning they were pre-clinical compounds. This was confirmed through the answer from ChEMBL FAQ link, a max phase of “null” compound means:
-
-
“Preclinical (NULL): preclinical compounds with bioactivity data e.g. is a preclinical compound with bioactivity data that has been extracted from scientific literature. However, the sources of drug and clinical candidate drug information in ChEMBL do not show that this compound has reached clinical trials and therefore the max_phase is set to null.”
-
-
Again, setting up the features for the testing dataset.
Because of the small amount of training data, this might hint at using an ensemble approach in the future, where model averaging would be derived from a bunch of tree models rather than using a single tree model, which was what I did here. The reason I started with one tree was because it was no point in building a multiple-tree model if one had no clue about how one tree was built. To learn as much as possible, I had to dissect the multiple-tree version first to focus on one tree at a time.
-
One thing I’d like to mention was that rivastigmine was not included in the training dataset because it was actually not a pure AChE inhibitor (as it was also a butyrylcholinesterase (BChE) inhibitor), since my focus was purely on AChE at this time, this particular drug was unfortunately excluded. However, I did make sure the other two drugs (donepezil and galantamine) were included in the training dataset. One possible thing to do in the future if I want to improve this was to add BChE as another protein target and perhaps add this as an additional dataset towards the model.
-
As described in the subsection of “Estimate experimental errors”, there were experimental errors of 3-fold, 5-fold and 10-fold estimated based on the provided pKi data. With the prediction model used in this post, the estimated experimental errors would need to be taken into consideration, particularly at the time when the model was being investigated during the model evaluation and validation step (however due to the length of series 2.1 posts, I decided not to add this step yet, but would try to look at this later in the multiple tree model series if this applies).
-
A bit about the last decision tree plot, tentatively clogp (calculated partition coefficient) might be the crucial molecular feature in deciding whether a molecule might be closer to being an AChE inhibitor. Other important molecular features also included the number of aromatic rings, molecular weights, solvent accessible surface area and others (I’ve set the random state to 1 for now, so hopefully the result will be reproducible as I realised my old code without it always generated different tree plots, then all of the sudden I remembered that I forgot to set the random state of the estimator, so this was added).
-
Since the type of AChE inhibitors was not the focus of this series, I won’t go into details about which value of pKi or Ki would lead to the ideal AChE inhibitor (the well-known Cheng-Prusoff equation (Cheng and Prusoff 1973) might also lead to many discussions about Ki and IC50 values). This is because there are at least two types of AChE inhibitors available - reversible and irreversible (Colovic et al. 2013). Donepezil, galantamine and rivastigmine mentioned previously are the commonly known reversible AChE inhibitors. The irreversible type, as the name suggested, is usually used as insecticides or nerve agents. Another reason is that I didn’t go into details checking all of the identities for the 10 max phase 4 compounds used in the training set, as I only really made sure that donepezil and galantamine were included in the 10 molecules. If I were to re-model again purely on reversible AChE inhibitors targeting dementia or Alzheimer’s disease, I think I had to be quite sure of what I was training the model with, i.e. excluding irreversible AChE inhibitors from the training set.
-
However, if our aim was to only find novel AChE inhibitors in a general manner, one of the ways to check post-model building would be to re-run the dataframe again on compounds with max phase as null, including the molecular feature names to find out which compounds were at the predicted threshold cut-off values to see if their corresponding pKi values (note: these preclinical compounds had Ki values extracted from literature sources etc.) would match the predicted ones. One caveat of this method was that there might be pre-existing experimental errors in all the obtained and recorded Ki values, so this might not confirm that the model was truly a good reflection of the real-life scenario. Therefore, at most, this would probably add a very small value during the model evaluation phase.
-
The best way would be to test all of these compounds in the same experimental set-ups, through same experimental steps, and in the same laboratory to find out their respective Ki (or pKi) values. However, this was most likely not very feasible due to various real-life restrictions (the availability of financial and staffing resources). The most likely outcome might be to choose a selected group of compound candidates with the highest possibilities to proceed in the drug discovery pipeline based on past experimental, ML and clinical experiences, and then test them in the ways mentioned here.
-
I also came across a blog post about calculating the prediction intervals of ML prediction models (which mentioned the MAPIE package), but I didn’t quite get time to look into this package yet, and from what I have read in its repository link, it potentially could be quite useful for classification, regression and time-series models.
-
-
-
-
Final words
-
I didn’t think a definite conclusion could be drawn here, as this was only purely from one very simple and single decision tree, so I have named this last part as “final words”, as I felt if I didn’t stop here, this post or series of posts could go on forever or as long it could. The main thing here was to fully understand how one single decision tree was constructed based on hopefully reasonable-ish data (still not the best as I could not rule out all the noises from the data), and then to view the tree visually in different styles of plots. It was also important to understand how this was a white-box ML approach with clear features or descriptions shown to trace where the tree would branch off to reach different final outcomes or targets. This series was really a preamble for the multiple-tree models e.g. random forest and boosted trees, as I have bravely planned to do a series of posts on tree models due to my interests in them, so that might take a while, slowly but hopefully surely.
-
-
-
-
Acknowledgements
-
I’d like to thank all the authors for all the open-source packages used in the series 2.1 posts. I’d also like to thank all the authors of all the blog posts mentioned in this series as well since I’ve learnt a lot from them too.
-
-
-
-
-
-
References
-
-Brown, Scott P., Steven W. Muchmore, and Philip J. Hajduk. 2009. “Healthy Skepticism: Assessing Realistic Model Performance.”Drug Discovery Today 14 (7-8): 420–27. https://doi.org/10.1016/j.drudis.2009.01.012.
-
-
-Cheng, Yung-Chi, and William H. Prusoff. 1973. “Relationship Between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction.”Biochemical Pharmacology 22 (23): 3099–3108. https://doi.org/10.1016/0006-2952(73)90196-2.
-
-
-Colovic, Mirjana B., Danijela Z. Krstic, Tamara D. Lazarevic-Pasti, Aleksandra M. Bondzic, and Vesna M. Vasic. 2013. “Acetylcholinesterase Inhibitors: Pharmacology and Toxicology.”Current Neuropharmacology 11 (3): 315–35. https://doi.org/10.2174/1570159x11311030006.
-
-
]]>
- Machine learning projects
- Tree models
- Pandas
- Scikit-learn
- ChEMBL database
- Python
- https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/3_model_build.html
- Mon, 18 Sep 2023 12:00:00 GMT
-
-
-
- Molecular visualisation (Molviz) web application
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/15_Molviz/Molviz.html
- The final deployed app is on Shinyapps.io:
-
-
Link: here or please visit https://jhylin.shinyapps.io/molviz_app/
-
Code: here or please visit https://github.com/jhylin/Molviz_app
-
-
-
-
Background - how the app started
-
Originally I had an idea of incorporating mols2grid library within Shiny for Python web app framework (after seeing an example of a similar app in Streamlit previously). So I worked on a few ideas, but obviously mols2grid was designed to work inside Jupyter Notebook/Lab and Shiny for Python was only out of alpha at that stage so things were still being developed. After a few trials, unfortunately mols2grid wasn’t directly compatible with the Shiny for Python framework at that time (I even wrote a small story about it as a comment to an issue).
-
I then went away to work on another project on molecular scaffolds and left this mini project aside. However, recently I had another idea of trying to build a Shiny for Python app from the scratch (with a focus on cheminformatics or chemical information), so that users in relevant fields can view and save 2D images of small molecules in a web browser environment instead of only inside a Jupyter Notebook/Lab. I also thought to place the Shiny for Python framework to test if it was being used in a more intensive area such as chemistry and drug discovery.
-
Another reason that have triggered this side project was that I came across a comment from an old RDKit blog post from someone asking about how to save compound image as a PNG1 file, since SVG2 version was hard to convert etc. (or something along that line). I thought it should be possible, and this should not be only limited to Jupyter environments only (thinking of people not doing coding at all…), so here we are.
-
-
-
-
About each version of the app
-
I’ll try to explain what each version of the app_x.py script entails, as there are currently several different versions of them inside the repository. The final version is the one called “app.py”, named this way so that it’ll be recognised by rsconnect/Shinyapps.io when deploying the app. By providing some explanations below should also show that it was quite a process to arrive at the final “app.py”, it wasn’t built within a day for sure (at least for me).
-
-
-
app_molviz_v1.py
-
This was the first version that purely provided the ability to show 2D images of the molecules via selecting their corresponding index numbers. The libraries used appeared less aligned and a few tests were run below (some of them commented out during trials). This was the one that I’ve figured out how to make the image appeared in the app.
-
-
-
-
app_molviz_v2.py
-
For the second version, I started thinking about how I would present the app in a simple layout for the end users. The backbone code to support image generations was by using rdkit.Chem.Draw package’s MolToImage() module, which normally returns a PIL3 image object, and also supports atom and bond highlighting. Another useful module that I’ve tried was MolToFile() within the same package, which would generate and save a PNG file for a specified molecule from the dataframe.
-
I then took a bit more time to familiarise myself with some basic PIL image manipulations, and used online resources to formulate code to merge separate PNG images into one table grid-like image - potentially may be useful for substructural or R-group comparisons.
-
I have also added the interactive data table at the end to see how it would fit in with the rest of the app.
-
-
-
-
app_molviz_v3.py
-
The third version mainly dealt with how to segregate and differentiate between highlighting or non-highlighting and also with or without index numbers showing for the compounds in the images. I’ve tried to use a different code for atom labelling this time with thanks to this link. However, there was always an issue of not being able to flip back from with index to without index, since the atom labelling code itself somehow overflows its effect to the rest after labelling the atom indices (presumably this atom labelling code would work great in a Jupyter notebook scenario).
-
-
-
-
app_molviz_v4.py & app_molviz_v5.py
-
Both version 4 and 5 were where I’ve tested using “atomNote” (numbers appear beside atoms) instead of “atomLabel” (numbers replaces atoms directly in structures) to label atoms in molecular structures.
-
An example of the atom labelling code would look like this (replace ‘atomNote’ with ‘atomLabel’ to get different labelling effect):
-
```{python}
-for atom in mols[input.mol()].GetAtoms():
- atom.SetProp('atomNote', str(atom.GetIdx()))
-```
-
I’ve also started adding introductory texts for the app and edited the layout a bit more.
-
-
-
-
app_molviz_v6_hf.py
-
This was basically the final version of the app, but with code edited to attempt to deploy the app on HuggingFace. The main difference I was testing was on how to store the saved images as Docker was new to me at the time, and then while I was thinking about changing the Dockerfile, there was actually another problem in relation to the cairosvg code. Because of this, I then placed this deployment on hold in order to buy more time to figure out code, and also to try Shinyapps.io to see if this could be deployed.
-
-
-
-
app_molviz_v6.py or app.py
-
This was the last version and was the version used to deploy the app on Shinyapps.io. I had to rename the file as mentioned previously to “app.py” so that the Shinyapps.io servers would recognise this Python script as the script to run the app (otherwise it wouldn’t be deployed successfully, this took me a few tries and to read the log file to figure this out). So it was saved as a separate file, and for any latest text changes in the app I would refer to app.py as the most current app file.
-
The biggest code change was that I ended up not using the MolToImage() or MolToFile() modules, but rather I used rdMolDraw2D module from rdkit.Chem.Draw package. The reason being I’ve noticed the image resolutions weren’t great for the previously used modules (Jupyter notebook environments should not have this problem, as you could simply switch on this line of code by setting it to true like this, IPythonConsole.ipython_useSVG = True). So I resorted to other means and came across this useful link to generate images with better resolutions, and introduced the cairosvg library.
-
So the code was changed and would now use rdMolDraw2D.MolDraw2DSVG() first and add on addAtomIndices from drawOptions() and also DrawMolecule() to highlight substructures. The SVG generated would then be converted to PNG via cairosvg library. The end result produced slightly better image resolutions. Although I’ve found for more structurally complexed molecules, the image size would really need to be quite large to be in the high resolution zone. For compounds with simpler structures, this seemed to be much less of a problem. This was also why I had to have these PNG images blown up this large in the app, to cater for the image resolution aspect.
-
-
-
-
-
Other files
-
-
code_test.py
-
I’m not exactly sure how other data scientists/developers work, but for me since I came from a completely different background and training, I’m used to plan, set up and do experiments to test things I’d like to try, and see where the results will lead me to. So for this in a virtual computer setting, I used the “code_tests.py” to test a lot of different code.
-
If you go into this file, you’ll likely see a lot of RDKit code trials, and I have had a lot of fun doing this since I got to see results straight away when running the code, and learn new code and functions that way. If the end result was not the one I’ve intended, I would go on short journeys to look for answers (surprisingly I didn’t use any generative AI chatbots actually), it would be chosen intuitively as I searched online, but for this particular project, a lot of it was from past RDKit blogs, StackOverflow and random snippets I came across that have given me ideas about solving issues I came across.
-
-
-
-
app_table.py & app_itables.py
-
These two files were trials for incorporating dataframe inside a web app. The difference was that app_table.py was just a data table sitting inside the app, without any other particular features, while app_itables.py utilised a different package called itables, which provided an interactive feature to search for data in the table. The previous post on data source used for this app was presented as an interactive data table embedded inside a Quarto document, the same principle would also apply for a table inside a Jupyter notebook environment.
-
-
-
-
app_sample.py
-
This file was provided by Posit (formerly known as RStudio) from their Python for Shiny app template in HuggingFace as an example script for an app.
-
-
-
-
-
Features of the app
-
There are three main features for this app which allows viewing, saving4 and highlighting substructures of target molecules as PNG image files. I’m contemplating about adding a download feature for image file saving on the deployed app version but because I’m currently using the free version of Shinyapps.io with limited amount of data available, this may be unlikely (also because the app is more of a demonstration really as the focus is not to provide particular data/image downloads).
-
-
-
-
App deployment
-
There were two places I’ve tried so far, which were HuggingFace and Shinyapps.io. As mentioned briefly earlier under the subsection of “app_molviz_v6_hf.py”, it turned out cairosvg code didn’t quite play out as expected. I have so far not returned to fix this yet on HuggingFace, since I’ve managed to deploy the app on Shinyapps.io. I had a feeling I might need to revert back to the older code version with poorer image resolutions, so that was also another reason why I haven’t fixed it yet as I’d prefer to keep the better resolution one (unless someone has better ideas out there).
-
However, deploying the app to Shinyapps.io also wasn’t a smooth ride as well, there were some problems initially. The very first problem I got was being told by the error message that rsconnect-python was only compatible with Python version 3.7, 3.8, 3.9 and 3.10 only. I did some information digging in the Posit community forum, and I think several people mentioned using 3.9 without any problems to deploy their apps. Python version 3.11 definitely did not work at all so please avoid for now if anyone would like to try using Shiny for Python app (unless updated by rsconnect-Python in the future).
-
So I think the ideal app building workflow might be like this:
-
Note: all code examples below are to be run in the command line interface
-
-
Refer to this link provided by Shiny for Python, which details about how to set up the working directory, download the Shiny package and create a virtual environment
-
When creating the virtual environment, use venv which was already built-in within Python (and also as suggested by the Shiny for Python link) and set it to a compatible Python version.
-
-
```{python}
-# To create a venv with a specific Python version e.g. Python 3.9
-python3.9-m venv my_venv_name
-
-# Activate the created venv
-source my_venv_name/bin/activate
-```
-
-
If you’ve accidentally set it to Python 3.11 (like what I did), just deactivate the old venv and re-create another one by using the code above. The code below can be used to deactivate the venv set up in the first place.
-
-
```{python}
-# Deactivate the old venv
-deactivate
-```
-
-
If you had to set up a new venv with a new Python version, and did not want to re-add/install all the packages or libraries used in the older version, save all the pip packages like this code below as a requirements.txt file.
-
-
```{python}
-pip freeze > requirements.txt
-```
-
-
Once the requirements.txt was saved and after the new venv was set up and activated, install all the saved packages used to run the app by using this code.
-
-
```{python}
-pip freeze -r requirements.txt
-```
-
-
Start coding for your app and have fun - don’t forget to save and push the files to your repository.
-
To deploy to Shinyapps.io, follow this link, which explains all the steps. One thing I would like to remind again here is to make sure the app script (i.e. the one with data source, user interface and server code) was saved as “app.py”, so that the rsconnect-python server will recognise it and be able to deploy it to the cloud environment.
-
-
-
-
-
Further improvements of the app
-
There are of course a few things I think could be done to make the app better.
-
-
It may be useful to add a download option as mentioned previously, but for demonstration purpose, I’m leaving it as a “View” only for now, unless I get comments from readers that they’d like to try this. For localhost version, the saving image function should work with files saved to the working directory.
-
It may be even better to use SMARTS5 or SMILES for highlighting compound substructures actually (atom and bond numbering can be a bit tricky, I’ve tried the app myself, and it might not be as straight forward). I’m using atom indices here since I’m using a specific code in RDKit, but perhaps more experienced users for RDKit will know how to make code alterations etc.
-
The app layout could be further optimised for aesthetics e.g. interactive data table could be placed at a different location, and potentially the data table could contain other data such as compound bioassay results to really fit in the structure-activity relationship exploring task.
-
-
-
-
-
Final words
-
The whole idea behind this side project was to show that interested users could use this web app framework to build an interactive app by using their own data. Other useful web app frameworks are also available out there and could potentially be more or equally useful as this one (I’m simply testing out Python for Shiny here since it’s relatively new). In a drug discovery and development setting, this could be useful to make non-coding members understand where the computer side was trying to do, and to possibly assist them during their lab workflows, hoping to add some convenience at least.
This is currently limited to localhost version if running the app.py in IDE such as VS Code, where the saved files can be located in the working directory. The deployed version on Shinyapps.io currently only allow image viewing and structure highlighting only.↩︎
-
SMILES arbitrary target specification↩︎
-
-
]]>
- Python
- Datamol
- Shiny
- Pandas
- Polars
- itables
- https://jhylin.github.io/Data_in_life_blog/posts/15_Molviz/Molviz.html
- Wed, 09 Aug 2023 12:00:00 GMT
-
-
- Molecular visualisation (Molviz) web application
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/15_Molviz/itables.html
-
-
Introduction
-
This time I’m trying to build a web application in the hope to contribute my two cents towards minimising the gap between computational and laboratory sides in a drug discovery (or chemistry-related work) setting. There are definitely many other useful web applications available out there for similar uses, but I guess each one has its own uniqueness and special features.
-
For this app, it is mostly aimed at lab chemists who do not use any computer programming code at all in their work, and would like to quickly view compounds in the lab while working, and also to be able to highlight compound substructures during discussions or brainstorming for ideas during compound synthesis. Importantly, this app can exist outside a Jupyter notebook environment with internet required to access the app.
-
This is also the first part prior to the next post which will showcase the actual app. This part mainly involves some data cleaning but not as a major focus for this post. This is not to say that data cleaning is not important, but instead they are fundamental to any work involving data in order to ensure reasonable data quality, which can then potentially influence decisions or results. I have also collapsed the code sections below to make the post easier to read (to access code used for each section, click on the “Code” links).
-
-
-
-
Code and explanations
-
It was actually surprisingly simple for this first part when I did it - building the interactive table. I came across this on LinkedIn on a random day for a post about itables being integrated with Shiny for Python plus Quarto. It came at the right time because I was actually trying to build this app. I quickly thought about incorporating it with the rest of the app so that users could refer back to the data quickly while visualising compound images. The code and explanations on building an interactive table for dataframes in Pandas and Polars were provided below.
-
To install itables, visit here for instructions and also for other information about supported notebook editors.
-
-
-Code
-
# Import dataframe libraries
-import pandas as pd
-import polars as pl
-
-# Import Datamol
-import datamol as dm
-
-# Import itables
-from itables import init_notebook_mode, show
-init_notebook_mode(all_interactive=True)
-
-
-# Option 1: Reading df_ai.csv as a pandas dataframe
-#df = pd.read_csv("df_ai.csv")
-#df.head
-
-# Option 2: Reading df_ai.csv as a polars dataframe
-df = pl.read_csv("df_ai.csv")
-#df.head()
-
-
-# Below was the code I used in my last post to fix the missing SMILES for neomycin
-# - the version below was edited due to recent updates in Polars
-# Canonical SMILES for neomycin was extracted from PubChem
-# (https://pubchem.ncbi.nlm.nih.gov/compound/Neomycin)
-
-df = df.with_columns([
- pl.when(pl.col("Smiles").str.lengths() ==0)
- .then(pl.lit("C1C(C(C(C(C1N)OC2C(C(C(C(O2)CN)O)O)N)OC3C(C(C(O3)CO)OC4C(C(C(C(O4)CN)O)O)N)O)O)N"))
- .otherwise(pl.col("Smiles"))
- .keep_name()
-])
-
-#df.head()
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
Polars dataframe library was designed without the index in mind (which is different to Pandas), therefore the itables library did not work on my specific polars dataframe which required an index column to show (note: all other Polars dataframes should work fine with itables without the index column!).
-
However to show row counts in Polars dataframes, we could use with_row_count() that starts the index from 0, and this would show up in a Jupyter environment as usual. A small code example would be like this below.
-
-
-Code
-
# Uncomment below to run
-#df = df.with_row_count()
-
-
-
Then I converted the Polars dataframe into a Pandas one (this could be completely avoided if you started with Pandas actually).
-
-
-Code
-
df = df.to_pandas()
-
-
-
Then I added Datamol’s “_preprocess” function to convert SMILES1 into other molecular representations such as standardised SMILES (pre-processed and cleaned SMILES), SELFIES2, InChI3, InChI keys - just to provide extra information for further uses if needed. The standardised SMILES generated here would then be used for generating the molecule images later (in part 2).
# Saving cleaned df_ai.csv as a new .csv file (for app_image_x.py - script to build the web app)
-# df = pl.from_pandas(df)
-# df.write_csv("df_ai_cleaned.csv", sep = ",")
-
-
-
-
-
Options for app deployment
-
Since I had a lot of fun deploying my previous app in Shinylive last time, I thought I might try the same this time - deploying the Molviz app as a Shinylive app in Quarto. However, it didn’t work as expected, with reason being that RDKit wasn’t written in pure Python (it was written in Python and C++), so there wasn’t a pure Python wheel file available in PyPi - this link may provide some answers relating to this. Essentially, packages or libraries used in the app will need to be compatible with Pyodide in order for the Shinylive app to work. So, the most likely option to deploy this app now would be in Shinyapps.io or HuggingFace as I read about it recently.
-
-
-
-
Next post
-
Code and explanations for the actual Molviz app will be detailed in the next post. To access full code and files used for now, please visit this repository link.
-
-
-
-
-
-
Footnotes
-
-
-
Simplified Molecular Input Line Entry Systems↩︎
-
SELF-referencIng Embedded Strings↩︎
-
International Chemical Identifier↩︎
-
-
]]>
- Python
- Datamol
- Pandas
- Polars
- itables
- https://jhylin.github.io/Data_in_life_blog/posts/15_Molviz/itables.html
- Wed, 09 Aug 2023 12:00:00 GMT
-
-
- Working with scaffolds in small molecules
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/14_Scaffolds_in_small_molecules/chembl_anti-inf_data_prep_current.html
-
-
Features in post
-
This post will mainly be about the following:
-
-
Pre-process and standardise compounds (e.g. converting SMILES1 into SELFIES2 and other forms)
-
Obtain scaffolds for compounds
-
Align scaffolds of compounds
-
Query target scaffolds against a dataframe of compounds:
-
-
Function for saving multiple SMILES in .smi file
-
Function for converting .smi file into a list to query and match scaffolds of interests
-
Identify any similarities or differences in target compound of interest against other compounds in a dataframe
-
-
-
-
-
-
Quick words
-
I’ve always wanted to shorten my posts to a more readable length, but it was proven to be hard again, as this post was much longer than expected. Page content links are available on the right-hand side if needing to jump to sections for quick reads.
-
-
-
-
Key question to answer
-
Will the scaffold of compound 3 (compound of interest) be similar to the scaffolds of any approved anti-infectives in ChEMBL database?
-
-
-
-
Import libraries
-
The following libraries were used in this post.
-
-
import polars as pl
-import pandas as pd
-import datamol as dm
-import mols2grid
-
-from rdkit import Chem
-from rdkit.Chem import AllChem
-from rdkit.Chem.rdmolfiles import SmilesWriter, SmilesMolSupplier
-
-# Following library was modified & adapted from
-# Patrick Walters' tutorial on "Identifying Scaffolds"
-# - links provided in "scaffold_finder library" section under
-# the subheading of "Combining ChEMBL anti-infectives and FtsZ compounds"
-from scaffold_finder_test import find_scaffolds, get_molecules_with_scaffold
-
-
-
-
-
ChEMBL anti-infectives
-
-
Data cleaning
-
The dataset used was extracted from ChEMBL database, with a focus on the anti-infectives.
# Uncomment below if requiring a quick overview on all column names,
-# first ten variables in each column and each column data type
-#print(df_ai.glimpse())
-
-
Under the “Availability Type” column, there were a few different availabilities for each anti-bacterial such as, “Discontinued”, “Withdrawn”, “Unknown” and “Prescription Only”.
-
-
df_ai.groupby("Availability Type").count()
-
-
-
-
-shape: (4, 2)
-
-
-
-
-Availability Type
-
-
-count
-
-
-
-
-str
-
-
-u32
-
-
-
-
-
-
-"Discontinued"
-
-
-36
-
-
-
-
-"Withdrawn"
-
-
-7
-
-
-
-
-"Unknown"
-
-
-29
-
-
-
-
-"Prescription O...
-
-
-72
-
-
-
-
-
-
-
-
I only wanted to choose the “Prescription Only” ones, so the following filter condition was applied to the dataframe.
In preparation for possible future work on building machine learning models on this line of work, I looked into Datamol’s function on pre-processing molecules (shown in the next section), as it involved converting SMILES strings into SELFIES, which were considered to be more robust than SMILES.
-
However, I kept running into an error, with the error message showing the SMILES column was empty. After a few tries I realised that I’ve actually forgotten to check whether there were any missing SMILES in the column. So here I’ve filtered the SMILES column to look for any missing SMILES
-
-
df_ai_rx.filter(pl.col("Smiles") =="")
-
-
-
-
-shape: (1, 29)
-
-
-
-
-Parent Molecule
-
-
-Name
-
-
-Synonyms
-
-
-Research Codes
-
-
-Phase
-
-
-Drug Applicants
-
-
-USAN Stem
-
-
-USAN Year
-
-
-USAN Definition
-
-
-USAN Stem - Substem
-
-
-First Approval
-
-
-ATC Codes
-
-
-Level 4 ATC Codes
-
-
-Level 3 ATC Codes
-
-
-Level 2 ATC Codes
-
-
-Level 1 ATC Codes
-
-
-Indication Class
-
-
-Patent
-
-
-Drug Type
-
-
-Passes Rule of Five
-
-
-First In Class
-
-
-Chirality
-
-
-Prodrug
-
-
-Oral
-
-
-Parenteral
-
-
-Topical
-
-
-Black Box
-
-
-Availability Type
-
-
-Smiles
-
-
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-f64
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-i64
-
-
-i64
-
-
-str
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-str
-
-
-str
-
-
-
-
-
-
-"CHEMBL3989751"
-
-
-"NEOMYCIN"
-
-
-"FRADIOMYCIN|KA...
-
-
-""
-
-
-4.0
-
-
-"Bayer Pharmace...
-
-
-"'-mycin'"
-
-
-"1966"
-
-
-"antibiotics (S...
-
-
-"'-mycin(-mycin...
-
-
-"1957"
-
-
-"R02AB01 | S01A...
-
-
-"R02AB - Antibi...
-
-
-"R02A - THROAT ...
-
-
-"R02 - THROAT P...
-
-
-"R - RESPIRATOR...
-
-
-""
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-1
-
-
-0
-
-
-1
-
-
-1
-
-
-"Prescription O...
-
-
-""
-
-
-
-
-
-
-
-
Neomycin was the only compound found to have no SMILES recorded. To fix this error, I then used the “when-then-otherwise” expression in Polars again (used in previous post) to replace the empty string in the dataframe. A code example below was kindly adapted from StackOverflow from this link, and code example as shown below.
# Canonical SMILES for neomycin was extracted from PubChem
-# (https://pubchem.ncbi.nlm.nih.gov/compound/Neomycin)
-
-df_ai_rx = df_ai_rx.with_columns([
- pl.when(pl.col("Smiles").str.lengths() ==0)
- .then("C1C(C(C(C(C1N)OC2C(C(C(C(O2)CN)O)O)N)OC3C(C(C(O3)CO)OC4C(C(C(C(O4)CN)O)O)N)O)O)N")
- .otherwise(pl.col("Smiles"))
- .keep_name()
-])
-
-df_ai_rx
-
-
-
-
-shape: (72, 29)
-
-
-
-
-Parent Molecule
-
-
-Name
-
-
-Synonyms
-
-
-Research Codes
-
-
-Phase
-
-
-Drug Applicants
-
-
-USAN Stem
-
-
-USAN Year
-
-
-USAN Definition
-
-
-USAN Stem - Substem
-
-
-First Approval
-
-
-ATC Codes
-
-
-Level 4 ATC Codes
-
-
-Level 3 ATC Codes
-
-
-Level 2 ATC Codes
-
-
-Level 1 ATC Codes
-
-
-Indication Class
-
-
-Patent
-
-
-Drug Type
-
-
-Passes Rule of Five
-
-
-First In Class
-
-
-Chirality
-
-
-Prodrug
-
-
-Oral
-
-
-Parenteral
-
-
-Topical
-
-
-Black Box
-
-
-Availability Type
-
-
-Smiles
-
-
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-f64
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-i64
-
-
-i64
-
-
-str
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-str
-
-
-str
-
-
-
-
-
-
-"CHEMBL186"
-
-
-"CEFEPIME"
-
-
-"BMY-28142|CEFE...
-
-
-"BMY-28142|J01D...
-
-
-4.0
-
-
-"Samson Medical...
-
-
-"'cef-'"
-
-
-"1987"
-
-
-"cephalosporins...
-
-
-"'cef-(cef-)'"
-
-
-"1996"
-
-
-"J01DE01"
-
-
-"J01DE - Fourth...
-
-
-"J01D - OTHER B...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial"
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"CO/N=C(\C(=O)N...
-
-
-
-
-"CHEMBL2364632"
-
-
-"SARECYCLINE"
-
-
-"P-005672|P0056...
-
-
-"P-005672|P0056...
-
-
-4.0
-
-
-"Almirall Llc"
-
-
-"'-cycline'"
-
-
-"2012"
-
-
-"antibiotics (t...
-
-
-"'-cycline(-cyc...
-
-
-"2018"
-
-
-"J01AA14"
-
-
-"J01AA - Tetrac...
-
-
-"J01A - TETRACY...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-""
-
-
-"US-8318706-B2"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"CON(C)Cc1ccc(O...
-
-
-
-
-"CHEMBL31"
-
-
-"GATIFLOXACIN"
-
-
-"AM-1155|BMS-20...
-
-
-"AM-1155|BMS-20...
-
-
-4.0
-
-
-"Apotex Inc|Bri...
-
-
-"'-oxacin'"
-
-
-"1997"
-
-
-"antibacterials...
-
-
-"'-oxacin(-oxac...
-
-
-"1999"
-
-
-"S01AE06 | J01M...
-
-
-"S01AE - Fluoro...
-
-
-"S01A - ANTIINF...
-
-
-"S01 - OPHTHALM...
-
-
-"S - SENSORY OR...
-
-
-"Antibacterial"
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Racemic Mixtur...
-
-
-0
-
-
-1
-
-
-1
-
-
-1
-
-
-0
-
-
-"Prescription O...
-
-
-"COc1c(N2CCNC(C...
-
-
-
-
-"CHEMBL3039597"
-
-
-"GENTAMICIN"
-
-
-"GENTAMICIN|GEN...
-
-
-"SCH-9724"
-
-
-4.0
-
-
-"Schering Corp ...
-
-
-"'-micin'"
-
-
-"1963"
-
-
-"antibiotics (M...
-
-
-"'-micin(-micin...
-
-
-"1970"
-
-
-"S01AA11 | S02A...
-
-
-"S01AA - Antibi...
-
-
-"S01A - ANTIINF...
-
-
-"S01 - OPHTHALM...
-
-
-"S - SENSORY OR...
-
-
-"Antibacterial"
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Unknown"
-
-
-0
-
-
-0
-
-
-1
-
-
-1
-
-
-1
-
-
-"Prescription O...
-
-
-"CNC(C)[C@@H]1C...
-
-
-
-
-"CHEMBL893"
-
-
-"DICLOXACILLIN"
-
-
-"BRL-1702|DICLO...
-
-
-"BRL-1702|R-134...
-
-
-4.0
-
-
-"Teva Pharmaceu...
-
-
-"'-cillin'"
-
-
-"1965"
-
-
-"penicillins"
-
-
-"'-cillin(-cill...
-
-
-"1968"
-
-
-"J01CF01"
-
-
-"J01CF - Beta-l...
-
-
-"J01C - BETA-LA...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial"
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"Cc1onc(-c2c(Cl...
-
-
-
-
-"CHEMBL1449"
-
-
-"TICARCILLIN"
-
-
-"TICARCILLIN|Ti...
-
-
-""
-
-
-4.0
-
-
-"Glaxosmithklin...
-
-
-"'-cillin'"
-
-
-"1973"
-
-
-"penicillins"
-
-
-"'-cillin(-cill...
-
-
-"1976"
-
-
-"J01CA13"
-
-
-"J01CA - Penici...
-
-
-"J01C - BETA-LA...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial"
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"CC1(C)S[C@@H]2...
-
-
-
-
-"CHEMBL1220"
-
-
-"TINIDAZOLE"
-
-
-"CP-12,574|CP-1...
-
-
-"CP-12,574|CP-1...
-
-
-4.0
-
-
-"Mission Pharma...
-
-
-"'-nidazole'"
-
-
-"1970"
-
-
-"antiprotozoal ...
-
-
-"'-nidazole(-ni...
-
-
-"2004"
-
-
-"P01AB02 | J01X...
-
-
-"P01AB - Nitroi...
-
-
-"P01A - AGENTS ...
-
-
-"P01 - ANTIPROT...
-
-
-"P - ANTIPARASI...
-
-
-"Antiprotozoal"
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Achiral Molecu...
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-1
-
-
-"Prescription O...
-
-
-"CCS(=O)(=O)CCn...
-
-
-
-
-"CHEMBL501122"
-
-
-"CEFTAROLINE FO...
-
-
-"CEFTAROLINE FO...
-
-
-"PPI-0903|TAK 5...
-
-
-4.0
-
-
-"Apotex Inc|All...
-
-
-"'cef-; fos-'"
-
-
-"2006"
-
-
-"cephalosporins...
-
-
-"'cef-(cef-); f...
-
-
-"2010"
-
-
-"J01DI02"
-
-
-"J01DI - Other ...
-
-
-"J01D - OTHER B...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-""
-
-
-"US-6417175-B1"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Single Stereoi...
-
-
-1
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"CCO/N=C(\C(=O)...
-
-
-
-
-"CHEMBL137"
-
-
-"METRONIDAZOLE"
-
-
-"ACEA|ANABACT|B...
-
-
-"BAY-5360|BAYER...
-
-
-4.0
-
-
-"Inforlife Sa|L...
-
-
-"'-nidazole'"
-
-
-"1962"
-
-
-"antiprotozoal ...
-
-
-"'-nidazole(-ni...
-
-
-"1963"
-
-
-"D06BX01 | P01A...
-
-
-"D06BX - Other ...
-
-
-"D06B - CHEMOTH...
-
-
-"D06 - ANTIBIOT...
-
-
-"D - DERMATOLOG...
-
-
-"Antibacterial,...
-
-
-"US-6881726-B2"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Achiral Molecu...
-
-
-0
-
-
-1
-
-
-1
-
-
-1
-
-
-1
-
-
-"Prescription O...
-
-
-"Cc1ncc([N+](=O...
-
-
-
-
-"CHEMBL376140"
-
-
-"TIGECYCLINE"
-
-
-"TIGECYCLINE|Ty...
-
-
-"WAY-GAR-936"
-
-
-4.0
-
-
-"Apotex Inc|Fre...
-
-
-"'-cycline'"
-
-
-"2002"
-
-
-"antibiotics (t...
-
-
-"'-cycline(-cyc...
-
-
-"2005"
-
-
-"J01AA12"
-
-
-"J01AA - Tetrac...
-
-
-"J01A - TETRACY...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-""
-
-
-"US-7879828-B2"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-0
-
-
-1
-
-
-0
-
-
-1
-
-
-"Prescription O...
-
-
-"CN(C)c1cc(NC(=...
-
-
-
-
-"CHEMBL1741"
-
-
-"CLARITHROMYCIN...
-
-
-"6-O-METHYLERYT...
-
-
-"A-56268|ABBOTT...
-
-
-4.0
-
-
-"Sunshine Lake ...
-
-
-"'-mycin'"
-
-
-"1988"
-
-
-"antibiotics (S...
-
-
-"'-mycin(-mycin...
-
-
-"1991"
-
-
-"J01FA09"
-
-
-"J01FA - Macrol...
-
-
-"J01F - MACROLI...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial"
-
-
-"US-7977488-B2"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"CC[C@H]1OC(=O)...
-
-
-
-
-"CHEMBL1747"
-
-
-"TOBRAMYCIN"
-
-
-"47663|Aktob|BE...
-
-
-"47663|NSC-1805...
-
-
-4.0
-
-
-"Igi Laboratori...
-
-
-"'-mycin'"
-
-
-"1972"
-
-
-"antibiotics (S...
-
-
-"'-mycin(-mycin...
-
-
-"1975"
-
-
-"J01GB01 | S01A...
-
-
-"J01GB - Other ...
-
-
-"J01G - AMINOGL...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial"
-
-
-"US-6987094-B2"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-0
-
-
-1
-
-
-1
-
-
-1
-
-
-"Prescription O...
-
-
-"NC[C@H]1O[C@H]...
-
-
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-...
-
-
-
-
-"CHEMBL404"
-
-
-"TAZOBACTAM"
-
-
-"CL 298,741|CL-...
-
-
-"CL 298,741|CL-...
-
-
-4.0
-
-
-"Wytells Pharma...
-
-
-"'-bactam'"
-
-
-"1989"
-
-
-"beta-lactamase...
-
-
-"'-bactam(-bact...
-
-
-"1993"
-
-
-"J01CG02"
-
-
-"J01CG - Beta-l...
-
-
-"J01C - BETA-LA...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Inhibitor (bet...
-
-
-"US-6900184-B2"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"C[C@]1(Cn2ccnn...
-
-
-
-
-"CHEMBL1689772"
-
-
-"OMADACYCLINE"
-
-
-"AMADACYCLINE|B...
-
-
-"BAY 73-6944|PT...
-
-
-4.0
-
-
-"Paratek Pharma...
-
-
-"'-cycline'"
-
-
-"2009"
-
-
-"antibiotics (t...
-
-
-"'-cycline(-cyc...
-
-
-"2018"
-
-
-"J01AA15"
-
-
-"J01AA - Tetrac...
-
-
-"J01A - TETRACY...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-""
-
-
-"US-7326696-B2"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"CN(C)c1cc(CNCC...
-
-
-
-
-"CHEMBL3989974"
-
-
-"CEFIDEROCOL"
-
-
-"CEFIDEROCOL|GS...
-
-
-"GSK2696266|S-6...
-
-
-4.0
-
-
-"Shionogi Inc"
-
-
-"'cef-'"
-
-
-"2017"
-
-
-"cephalosporins...
-
-
-"'cef-(cef-)'"
-
-
-"2019"
-
-
-"J01DI04"
-
-
-"J01DI - Other ...
-
-
-"J01D - OTHER B...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-""
-
-
-"US-9238657-B2"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"CC(C)(O/N=C(\C...
-
-
-
-
-"CHEMBL1435"
-
-
-"CEFAZOLIN"
-
-
-"CEFAZOLIN|CEPH...
-
-
-"J01DB04|SK&F-4...
-
-
-4.0
-
-
-"Glaxosmithklin...
-
-
-"'cef-'"
-
-
-"1972"
-
-
-"cephalosporins...
-
-
-"'cef-(cef-)'"
-
-
-"1973"
-
-
-"J01DB04"
-
-
-"J01DB - First-...
-
-
-"J01D - OTHER B...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial ...
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"Cc1nnc(SCC2=C(...
-
-
-
-
-"CHEMBL3989751"
-
-
-"NEOMYCIN"
-
-
-"FRADIOMYCIN|KA...
-
-
-""
-
-
-4.0
-
-
-"Bayer Pharmace...
-
-
-"'-mycin'"
-
-
-"1966"
-
-
-"antibiotics (S...
-
-
-"'-mycin(-mycin...
-
-
-"1957"
-
-
-"R02AB01 | S01A...
-
-
-"R02AB - Antibi...
-
-
-"R02A - THROAT ...
-
-
-"R02 - THROAT P...
-
-
-"R - RESPIRATOR...
-
-
-""
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-1
-
-
-0
-
-
-1
-
-
-1
-
-
-"Prescription O...
-
-
-"C1C(C(C(C(C1N)...
-
-
-
-
-"CHEMBL572"
-
-
-"NITROFURANTOIN...
-
-
-"BERKFURIN|CEDU...
-
-
-"NSC-2107|NSC-4...
-
-
-4.0
-
-
-"Sun Pharmaceut...
-
-
-"'-toin'"
-
-
-""
-
-
-"antiepileptics...
-
-
-"'-toin(-toin)'...
-
-
-"1953"
-
-
-"J01XE01 | J01X...
-
-
-"J01XE - Nitrof...
-
-
-"J01X - OTHER A...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial ...
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Achiral Molecu...
-
-
-0
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-"Prescription O...
-
-
-"O=C1CN(/N=C/c2...
-
-
-
-
-"CHEMBL44354"
-
-
-"CEFTAZIDIME"
-
-
-"CEFTAZIDIME|CE...
-
-
-"GR 20263|GR-20...
-
-
-4.0
-
-
-"Glaxosmithklin...
-
-
-"'cef-'"
-
-
-"1980"
-
-
-"cephalosporins...
-
-
-"'cef-(cef-)'"
-
-
-"1985"
-
-
-"J01DD02"
-
-
-"J01DD - Third-...
-
-
-"J01D - OTHER B...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial"
-
-
-"US-7112592-B2"
-
-
-"1:Synthetic Sm...
-
-
-0
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-0
-
-
-1
-
-
-0
-
-
-1
-
-
-"Prescription O...
-
-
-"CC(C)(O/N=C(\C...
-
-
-
-
-"CHEMBL29"
-
-
-"BENZYLPENICILL...
-
-
-"BENZYL PENICIL...
-
-
-"J01CE01|NSC-19...
-
-
-4.0
-
-
-"Hq Specialty P...
-
-
-"'-cillin'"
-
-
-""
-
-
-"penicillins"
-
-
-"'-cillin(-cill...
-
-
-"1947"
-
-
-"J01CE01 | S01A...
-
-
-"J01CE - Beta-l...
-
-
-"J01C - BETA-LA...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial"
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Single Stereoi...
-
-
-0
-
-
-1
-
-
-1
-
-
-0
-
-
-1
-
-
-"Prescription O...
-
-
-"CC1(C)S[C@@H]2...
-
-
-
-
-"CHEMBL8"
-
-
-"CIPROFLOXACIN"
-
-
-"BAY O 9867 FRE...
-
-
-"BAY O 9867 FRE...
-
-
-4.0
-
-
-"Inforlife Sa|A...
-
-
-"'-oxacin'"
-
-
-"1987"
-
-
-"antibacterials...
-
-
-"'-oxacin(-oxac...
-
-
-"1987"
-
-
-"J01MA02 | S03A...
-
-
-"J01MA - Fluoro...
-
-
-"J01M - QUINOLO...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial"
-
-
-"US-8318817-B2"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Achiral Molecu...
-
-
-0
-
-
-1
-
-
-1
-
-
-1
-
-
-1
-
-
-"Prescription O...
-
-
-"O=C(O)c1cn(C2C...
-
-
-
-
-"CHEMBL9"
-
-
-"NORFLOXACIN"
-
-
-"Baccidal|CHIBR...
-
-
-"MK-366|NSC-757...
-
-
-4.0
-
-
-"Merck Research...
-
-
-"'-oxacin'"
-
-
-"1984"
-
-
-"antibacterials...
-
-
-"'-oxacin(-oxac...
-
-
-"1986"
-
-
-"J01MA06 | S01A...
-
-
-"J01MA - Fluoro...
-
-
-"J01M - QUINOLO...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial"
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Achiral Molecu...
-
-
-0
-
-
-1
-
-
-0
-
-
-1
-
-
-1
-
-
-"Prescription O...
-
-
-"CCn1cc(C(=O)O)...
-
-
-
-
-"CHEMBL21"
-
-
-"SULFANILAMIDE"
-
-
-"ANILINE-P-SULF...
-
-
-"NSC-7618"
-
-
-4.0
-
-
-"Mylan Specialt...
-
-
-"'sulfa-'"
-
-
-""
-
-
-"antimicrobials...
-
-
-"'sulfa-(sulfa-...
-
-
-"1985"
-
-
-"D06BA05 | J01E...
-
-
-"D06BA - Sulfon...
-
-
-"D06B - CHEMOTH...
-
-
-"D06 - ANTIBIOT...
-
-
-"D - DERMATOLOG...
-
-
-""
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Achiral Molecu...
-
-
-0
-
-
-0
-
-
-0
-
-
-1
-
-
-0
-
-
-"Prescription O...
-
-
-"Nc1ccc(S(N)(=O...
-
-
-
-
-"CHEMBL4"
-
-
-"OFLOXACIN"
-
-
-"DL-8280|EXOCIN...
-
-
-"DL-8280|HOE 28...
-
-
-4.0
-
-
-"Bausch And Lom...
-
-
-"'-oxacin'"
-
-
-"1984"
-
-
-"antibacterials...
-
-
-"'-oxacin(-oxac...
-
-
-"1990"
-
-
-"J01MA01 | S02A...
-
-
-"J01MA - Fluoro...
-
-
-"J01M - QUINOLO...
-
-
-"J01 - ANTIBACT...
-
-
-"J - ANTIINFECT...
-
-
-"Antibacterial"
-
-
-"None"
-
-
-"1:Synthetic Sm...
-
-
-1
-
-
-0
-
-
-"Racemic Mixtur...
-
-
-0
-
-
-1
-
-
-1
-
-
-1
-
-
-1
-
-
-"Prescription O...
-
-
-"CC1COc2c(N3CCN...
-
-
-
-
-
-
-
-
-
# Keeping only selected columns with information needed for later use
-df_ai_rx = df_ai_rx.select(["Smiles", "Name", "USAN Definition", "Level 4 ATC Codes"])
-df_ai_rx.head()
-
-
-
-
-shape: (5, 4)
-
-
-
-
-Smiles
-
-
-Name
-
-
-USAN Definition
-
-
-Level 4 ATC Codes
-
-
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-
-
-
-
-"CO/N=C(\C(=O)N...
-
-
-"CEFEPIME"
-
-
-"cephalosporins...
-
-
-"J01DE - Fourth...
-
-
-
-
-"CON(C)Cc1ccc(O...
-
-
-"SARECYCLINE"
-
-
-"antibiotics (t...
-
-
-"J01AA - Tetrac...
-
-
-
-
-"COc1c(N2CCNC(C...
-
-
-"GATIFLOXACIN"
-
-
-"antibacterials...
-
-
-"S01AE - Fluoro...
-
-
-
-
-"CNC(C)[C@@H]1C...
-
-
-"GENTAMICIN"
-
-
-"antibiotics (M...
-
-
-"S01AA - Antibi...
-
-
-
-
-"Cc1onc(-c2c(Cl...
-
-
-"DICLOXACILLIN"
-
-
-"penicillins"
-
-
-"J01CF - Beta-l...
-
-
-
-
-
-
-
-
The “Smiles” column name was changed below to ensure _preprocess function would work since the parameter “smiles_column” in _preprocess function had “smiles” with lowercase “s” (this of course could be the other way round, where we could change the parameter name in the function instead - the column name and parameter name had to match for the function to work). The “Name” column was changed accordingly for similar reason.
I also wanted to change the all capitalised compound names into lowercases for the ease of reading.
-
-
# Convert all compounds to lowercases
-df_ai_rx = df_ai_rx.with_columns(pl.col("names").str.to_lowercase())
-df_ai_rx.head()
-
-
-
-
-shape: (5, 4)
-
-
-
-
-smiles
-
-
-names
-
-
-USAN Definition
-
-
-Level 4 ATC Codes
-
-
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-
-
-
-
-"CO/N=C(\C(=O)N...
-
-
-"cefepime"
-
-
-"cephalosporins...
-
-
-"J01DE - Fourth...
-
-
-
-
-"CON(C)Cc1ccc(O...
-
-
-"sarecycline"
-
-
-"antibiotics (t...
-
-
-"J01AA - Tetrac...
-
-
-
-
-"COc1c(N2CCNC(C...
-
-
-"gatifloxacin"
-
-
-"antibacterials...
-
-
-"S01AE - Fluoro...
-
-
-
-
-"CNC(C)[C@@H]1C...
-
-
-"gentamicin"
-
-
-"antibiotics (M...
-
-
-"S01AA - Antibi...
-
-
-
-
-"Cc1onc(-c2c(Cl...
-
-
-"dicloxacillin"
-
-
-"penicillins"
-
-
-"J01CF - Beta-l...
-
-
-
-
-
-
-
-
Since Datamol was built as a thin layer library on top of RDKit, which was really only compatible with Pandas, I added the following step to convert the dataframe into a Pandas one.
-
-
df_ai_pd = df_ai_rx.to_pandas()
-df_ai_pd.head()
-
-
-
-
-
-
-
-
-
smiles
-
names
-
USAN Definition
-
Level 4 ATC Codes
-
-
-
-
-
0
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
cefepime
-
cephalosporins
-
J01DE - Fourth-generation cephalosporins
-
-
-
1
-
CON(C)Cc1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C...
-
sarecycline
-
antibiotics (tetracycline derivatives)
-
J01AA - Tetracyclines
-
-
-
2
-
COc1c(N2CCNC(C)C2)c(F)cc2c(=O)c(C(=O)O)cn(C3CC...
-
gatifloxacin
-
antibacterials (quinolone derivatives)
-
S01AE - Fluoroquinolones | J01MA - Fluoroquino...
-
-
-
3
-
CNC(C)[C@@H]1CC[C@@H](N)[C@@H](O[C@H]2[C@H](O)...
-
gentamicin
-
antibiotics (Micromonospora strains)
-
S01AA - Antibiotics | S02AA - Antiinfectives |...
-
-
-
4
-
Cc1onc(-c2c(Cl)cccc2Cl)c1C(=O)N[C@@H]1C(=O)N2[...
-
dicloxacillin
-
penicillins
-
J01CF - Beta-lactamase resistant penicillins
-
-
-
-
-
-
-
-
# Check the dataframe has been converted from Polars to Pandas
-type(df_ai_pd)
-
-
pandas.core.frame.DataFrame
-
-
-
-
-
-
Pre-processing and standardising molecules
-
I have borrowed and adapted the _preprocess function from Datamol (link here), as shown below. One of the convenient features in this function was that it also included a conversion from “mol” (RDKit molecule) to SELFIES amongst several other common molecular representations such as InChI3 and SMILES.
The images generated below might be quite small to see or read clearly. I’ve tried to increase the molecule size (mol_size) and also reduce the column numbers, but it still appeared the same. However, if the code was run in say VS Code, the compound images would appear larger when increasing the mol_size.
-
-
# Grab all SMILES of the cleaned/pre-processed ChEMBL anti-infectives
-df_ai_sm = data_mol_clean["standard_smiles"]
-
-# Load a list of these molecules in SMILES
-# dm.to_mol() has sanitize = True set as default
-mol_ls = [dm.to_mol(smile) for smile in df_ai_sm]
-
-# Alternative way to convert dataframe into a list of mols (same as mol_ls)
-# mols = dm.from_df(df_name, smiles_column = "Smiles")
-
-# Add compound name for each 2D image
-legends_c =list(data_mol_clean["names"])
-
-# Convert the list of molecules into 2D images
-dm.to_image(mol_ls, n_cols =4, mol_size = (400, 400), legends = legends_c)
-
-
-
-
-
-
-
-
Extract scaffolds
-
-
# Extract Murcko scaffolds from mol_ls (ChEMBL anti-infectives)
-m_scaffolds = [dm.to_scaffold_murcko(mol) for mol in mol_ls]
-dm.to_image(m_scaffolds, mol_size = (400, 400), legends = legends_c)
-
-
-
-
-
-
-
-
-
Filamenting temperature-sensitive mutant Z (FtsZ) compounds
Before I started cleaning any data on FtsZ compounds, I found this useful website, OPSIN: Open Parser for Systematic IUPAC nomenclature, with this link to the journal paper as an acknowledgement of the work. I’ve managed to convert these 3 FtsZ compounds by using their IUPAC names, which were inputted into OPSIN, and converted into the corresponding InChI or SMILES strings.
-
After that, I started by converting the InChI of compound 1 into a RDKit molecule, which could be visualised in 2D below.
-
-
# Convert compound 1 to mol from InChI
-cpd1 = dm.from_inchi("InChI=1S/C22H20O4/c23-18-9-4-15(5-10-18)8-13-21(25)20-3-1-2-17(22(20)26)14-16-6-11-19(24)12-7-16/h4-14,20,23-24H,1-3H2/b13-8+,17-14+")
-cpd1
-
-
-
-
-
I then converted compound 2 using SMILES string instead.
# Grab all SMILES from cleaned FtsZ compound dataset
-df_ai_ftsz = data_cleaned["standard_smiles"]
-
-# Load a list of these molecules in SMILES
-mol_ftsz_list = [dm.to_mol(smile) for smile in df_ai_ftsz]
-
-# Add compound names for each 2D image of compounds
-legends =list(data_cleaned["names"])
-
-# Convert the list of molecules into 2D images
-dm.to_image(mol_ftsz_list, n_cols =5, mol_size = (400, 400), legends = legends)
-
-
-
-
-
-
-
-
Extract scaffolds
-
-
# Get Murcko scaffolds of FtsZ compounds
-m_ftsz_scaffolds = [dm.to_scaffold_murcko(mol) for mol in mol_ftsz_list]
-dm.to_image(m_ftsz_scaffolds, mol_size = (400, 400), legends = legends)
-
-
-
-
-
-
-
-
-
Combining ChEMBL anti-infectives and FtsZ compounds
-
-
Combining dataframes
-
In this part, I wanted to combine the two dataframes I had from above, since my next step was to compare the scaffolds between ChEMBL prescription-only anti-infectives and FtsZ compounds.
# Convert the standard SMILES into RDKit molecules
-mol_full = [dm.to_mol(smile) for smile in df_full]
-
-
-
-
-
Aligning all the scaffolds
-
Here, all the scaffolds from both dataframes of compounds were aligned by using Datamol’s auto_align_many(). The images of all the aligned molecules were generated in the end. The compound structures did re-align, but unfortunately it only showed up to a maximum of 50 compounds only (the maximum number of molecules to be shown in Datamol was set to 32 as default; this number was pushed up to and truncated at 50 in the warning message from RDKit when attempting to run a total of 75 compounds using the Datamol library, without looking further into other ways to alter this for now).
An attempt to combine Datamol’s auto_align_many() with mols2grid library was shown below. Unfortunately, the compounds did not re-align but all 75 compounds were shown in the grids.
-
-
mols2grid.display(aligned_list)
-
-
-
-
-
-
-
-
-
-
mols2grid library
-
Since I’ve started using mols2grid here, I thought to show an example of this library by combining all 75 compounds using the pre-processed standard SMILES in the grids with corresponding compound names. The resulting table provided a clear overview of all the compounds, with useful options to select or filter compounds. Obviously, other molecular properties or experimental results could be added into the table for other uses.
-
-
# Full dataset of 75 compounds
-mols2grid.display(full_data, smiles_col ="standard_smiles", subset = ["img", "mols2grid-id", "names"])
-
-
-
-
-
-
-
-
-
-
-
scaffold_finder library
-
Rather than only trying out Datamol only, I also thought to try out the scaffold_finder library after reading this Jupyter notebook by Patrick Walters. The GitHub repository of his other useful cheminformatics tutorials can be found here. His blog is here. Without surprises, this post was also inspired by this Jupyter notebook on “Identifying scaffolds in a set of molecules”, with some hope to expand on it a bit more.
-
Below were my notes on how to use this particular library.
-
Step 1: Add “mol” column to full_data dataframe (this was needed in order to use the functions from scaffold_finder library, which was also built based on RDKit)
Step 2: Change column names of “standard_smiles” to “SMILES” & “names” to “Name” to match with scaffold_finder library functions with set column names (or other way round, by changing the names in the function library)
-
-
# Note: New column name "SMILES" contains standardised SMILES (old column name as "standard_smiles")
-full_data = full_data.rename(columns = {"standard_smiles": "SMILES", "names": "Name"})
-full_data.head()
-
-
-
-
-
-
-
-
-
smiles
-
Name
-
USAN Definition
-
Level 4 ATC Codes
-
SMILES
-
selfies
-
inchi
-
inchikey
-
mol
-
-
-
-
-
0
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
cefepime
-
cephalosporins
-
J01DE - Fourth-generation cephalosporins
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
[C][O][/N][=C][Branch2][Ring2][#Branch2][\C][=...
-
InChI=1S/C19H24N6O5S2/c1-25(5-3-4-6-25)7-10-8-...
-
HVFLCNVBZFFHBT-ZKDACBOMSA-N
-
<rdkit.Chem.rdchem.Mol object at 0x134307680>
-
-
-
1
-
CON(C)Cc1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C...
-
sarecycline
-
antibiotics (tetracycline derivatives)
-
J01AA - Tetracyclines
-
CON(C)Cc1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C...
-
[C][O][N][Branch1][C][C][C][C][=C][C][=C][Bran...
-
InChI=1S/C24H29N3O8/c1-26(2)18-13-8-11-7-12-10...
-
PQJQFLNBMSCUSH-SBAJWEJLSA-N
-
<rdkit.Chem.rdchem.Mol object at 0x134306c00>
-
-
-
2
-
COc1c(N2CCNC(C)C2)c(F)cc2c(=O)c(C(=O)O)cn(C3CC...
-
gatifloxacin
-
antibacterials (quinolone derivatives)
-
S01AE - Fluoroquinolones | J01MA - Fluoroquino...
-
COc1c(N2CCNC(C)C2)c(F)cc2c(=O)c(C(=O)O)cn(C3CC...
-
[C][O][C][=C][Branch1][N][N][C][C][N][C][Branc...
-
InChI=1S/C19H22FN3O4/c1-10-8-22(6-5-21-10)16-1...
-
XUBOMFCQGDBHNK-UHFFFAOYSA-N
-
<rdkit.Chem.rdchem.Mol object at 0x133e729d0>
-
-
-
3
-
CNC(C)[C@@H]1CC[C@@H](N)[C@@H](O[C@H]2[C@H](O)...
-
gentamicin
-
antibiotics (Micromonospora strains)
-
S01AA - Antibiotics | S02AA - Antiinfectives |...
-
CNC(C)[C@@H]1CC[C@@H](N)[C@@H](O[C@H]2[C@H](O)...
-
[C][N][C][Branch1][C][C][C@@H1][C][C][C@@H1][B...
-
InChI=1S/C21H43N5O7.C20H41N5O7.C19H39N5O7/c1-9...
-
NPEFREDMMVQEPL-RWPARATISA-N
-
<rdkit.Chem.rdchem.Mol object at 0x133e72500>
-
-
-
4
-
Cc1onc(-c2c(Cl)cccc2Cl)c1C(=O)N[C@@H]1C(=O)N2[...
-
dicloxacillin
-
penicillins
-
J01CF - Beta-lactamase resistant penicillins
-
Cc1onc(-c2c(Cl)cccc2Cl)c1C(=O)N[C@@H]1C(=O)N2[...
-
[C][C][O][N][=C][Branch1][=N][C][=C][Branch1][...
-
InChI=1S/C19H17Cl2N3O5S/c1-7-10(12(23-29-7)11-...
-
YFAGHNZHGGCZAX-JKIFEVAISA-N
-
<rdkit.Chem.rdchem.Mol object at 0x133e719a0>
-
-
-
-
-
-
-
Step 3: Identify scaffolds
-
The find_scaffolds() function was kindly borrowed from the scaffold_finder library as mentioned above. The scaffold_finder_test.py was the modified version, as I’ve used a different dataset here.
-
-
mol_df, scaffold_df = find_scaffolds(full_data)
-
-
-
-
-
Below was a quick overview of the mol_df, showing scaffolds in SMILES, number of atoms, number of R groups, names of compounds and the standardised SMILES of the compounds.
-
-
mol_df
-
-
-
-
-
-
-
-
-
Scaffold
-
NumAtoms
-
NumRgroupgs
-
Name
-
SMILES
-
-
-
-
-
0
-
C[N+]1(CC2=C(C(=O)[O-])N3C(=O)[C@@H](NC(=O)/C(...
-
31
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
1
-
C[N+]1(CC2=CN3C(=O)[C@@H](NC(=O)C(=NO)c4csc(N)...
-
28
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
2
-
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=CCS[C@H]23)cs1
-
21
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
3
-
CC1=CN2C(=O)[C@@H](NC(=O)C(=NO)c3csc(N)n3)[C@H...
-
22
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
4
-
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=C(C[NH+]4CCC...
-
27
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
...
-
...
-
...
-
...
-
...
-
...
-
-
-
15
-
O=C(CCc1ccccc1)C1CCCC(Cc2ccccc2)C1=O
-
24
-
2
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
16
-
O=C1CCCCC1C(=O)CCc1ccc(O)cc1
-
18
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
19
-
CC1CCCC(C(=O)CCc2ccc(O)cc2)C1=O
-
19
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
21
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccccc2)C1=O
-
25
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
22
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
26
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
-
5320 rows × 5 columns
-
-
-
-
Again to have a quick look at the scaffolds of all 75 compounds, along with counts of each scaffold and number of atoms in each scaffold.
-
-
scaffold_df
-
-
-
-
-
-
-
-
-
Scaffold
-
Count
-
NumAtoms
-
-
-
-
-
1156
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
-
7
-
29
-
-
-
1101
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
6
-
30
-
-
-
173
-
CC(=O)N[C@@H]1C(=O)N2[C@@H]1SC(C)(C)[C@@H]2C(=O)O
-
5
-
17
-
-
-
174
-
CC(=O)N[C@@H]1C(=O)N2[C@@H]1SC(C)[C@@H]2C(=O)O
-
5
-
16
-
-
-
552
-
CC1(C)S[C@@H]2[C@H](NC=O)C(=O)N2[C@H]1C(=O)O
-
5
-
16
-
-
-
...
-
...
-
...
-
...
-
-
-
3684
-
Cc1nccn1C
-
1
-
7
-
-
-
4456
-
Nc1ccccc1
-
1
-
7
-
-
-
4775
-
O=P(O)(O)[C@@H]1CO1
-
1
-
7
-
-
-
3722
-
Cn1ccnc1
-
1
-
6
-
-
-
4807
-
c1ccccc1
-
1
-
6
-
-
-
-
4808 rows × 3 columns
-
-
-
-
Step 4: Display all scaffolds in mols2grid, which helped to identify the scaffold with the highest frequency (counts) of occurence in the dataset.
These were my sample datasets for later use in the section on “Reading and querying multiple scaffolds in SMILES strings”.
-
Below was the first test dataset on the top 2 scaffolds with highest frequency of appearance in the full dataframe.
-
-
# Scaffold of anti-infective with highest count
-count_top1_scaffold = scaffold_df.Scaffold.values[0]
-# Scaffold of anti-infective with the second highest count
-count_top2_scaffold = scaffold_df.Scaffold.values[1]
-
-
-
# Combine above scaffolds into a list
-count_top_scaffold =list((count_top1_scaffold, count_top2_scaffold))
-count_top_scaffold
Compound 3 was the compound found in the paper to have targeted the FtsZ proteins in Gram positive pathogens such as Streptococcus pneumoniae with more pronounced activities than its predecessor e.g. compound 1. So this section aimed to look into all of compound 3’s scaffolds.
-
-
# For ease of dataframe manipulation, decided to convert Pandas df into a Polars one (just my personal preference as I've used Polars more lately)
-# then filtered out all the scaffolds for compound 3 & saved it as an independent dataframe
-cpd3_scaffolds = pl.from_pandas(mol_df).filter(pl.col("Name") =="Compound_3")
-cpd3_scaffolds
-
-
-
-
-shape: (14, 5)
-
-
-
-
-Scaffold
-
-
-NumAtoms
-
-
-NumRgroupgs
-
-
-Name
-
-
-SMILES
-
-
-
-
-str
-
-
-i64
-
-
-i64
-
-
-str
-
-
-str
-
-
-
-
-
-
-"O=CC1CCCC(Cc2c...
-
-
-17
-
-
-1
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"O=CC1CCCC(Cc2c...
-
-
-16
-
-
-2
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"CC(=O)C1CCCC(C...
-
-
-18
-
-
-1
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"CC(=O)C1CCCC(C...
-
-
-17
-
-
-2
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"CCC(=O)C1CCCC(...
-
-
-19
-
-
-1
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"CCC(=O)C1CCCC(...
-
-
-18
-
-
-2
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"O=C(CCc1ccccc1...
-
-
-25
-
-
-1
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"O=C1CCCCC1C(=O...
-
-
-17
-
-
-2
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"CC1CCCC(C(=O)C...
-
-
-18
-
-
-2
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"O=C(CCc1ccccc1...
-
-
-24
-
-
-2
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"O=C1CCCCC1C(=O...
-
-
-18
-
-
-1
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"CC1CCCC(C(=O)C...
-
-
-19
-
-
-1
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"O=C(CCc1ccc(O)...
-
-
-25
-
-
-1
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-"O=C(CCc1ccc(O)...
-
-
-26
-
-
-1
-
-
-"Compound_3"
-
-
-"O=C(CCc1ccc(O)...
-
-
-
-
-
-
-
-
-
# Convert Polars df into a Pandas one
-# and use mols2grid to show the 2D images of compound 3 scaffolds
-# Total of 14 different scaffolds
-cpd3_scaffolds = cpd3_scaffolds.to_pandas()
-mols2grid.display(cpd3_scaffolds, smiles_col ="Scaffold")
-
-
-
-
-
-
-
-
-
Testing compound 3 scaffolds using scaffold_finder library
-
At this stage, I sort of had an idea of wanting to compare all 14 compound 3 scaffolds against all 75 molecules including ChEMBL-curated prescription-only anti-bacterials.
-
I tried the get_molecules_with_scaffold() function from scaffold_finder library but didn’t exactly get what I hoped to achieve. I played around a bit and noticed it was really designed for spotting a single target scaffold with highest counts in the data set. I was hoping to parse multiple scaffolds actually, or imagining there might be situations where we might want to do this.
-
I started trialling with one scaffold anyway as shown below on the get_molecule_with_scaffold() function from the scaffold_finder library.
-
-
# Trial single scaffold first
-scaffold_test = cpd3_scaffolds.Scaffold.values[0]
-scaffold_test
# Showing only compound 3 as a distinctive compound (no other molecules with similar scaffold)
-chem_mol_df
-
-
-
-
-
-
-
-
-
SMILES
-
Name
-
-
-
-
-
0
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
Compound_3
-
-
-
-
-
-
-
-
-
-
-
Reading and querying multiple scaffolds in SMILES strings
-
I also tried to tweak the get_molecule_with_scaffolds() function but realised it might be even better to write my own function to tailor to my need. Therefore, I wrote a small and simple function which would read and query multiple scaffolds of small molecules in SMILES string formats against a dataframe (with information showing scaffolds in SMILES, number of atoms, number of R groups, names of compounds and also the SMILES of the compounds).
-
At first, I started with reading all 14 scaffolds of compound 3 by using the values index method on cpd3_scaffolds dataframe that included all 14 scaffolds of compound 3.
-
-
# Trial feeding all 14 SMILES
-scaffold_cpd3_all = cpd3_scaffolds.Scaffold.values[:]
-scaffold_cpd3_all
Then I thought about how every time if we’d want to convert any molecules from SMILES to RDKit molecules, we really had to have a “mol” column set up, so that was what I did below.
Then perhaps I would place all of these compound 3 scaffolds into an object.
-
-
cpd3_mols = cpd3_scaffolds["mol"]
-cpd3_mols
-
-
0 <rdkit.Chem.rdchem.Mol object at 0x13580d3f0>
-1 <rdkit.Chem.rdchem.Mol object at 0x13580d4d0>
-2 <rdkit.Chem.rdchem.Mol object at 0x13580dd20>
-3 <rdkit.Chem.rdchem.Mol object at 0x13580e880>
-4 <rdkit.Chem.rdchem.Mol object at 0x13580d930>
-5 <rdkit.Chem.rdchem.Mol object at 0x13580ea40>
-6 <rdkit.Chem.rdchem.Mol object at 0x13580ca50>
-7 <rdkit.Chem.rdchem.Mol object at 0x13580fbc0>
-8 <rdkit.Chem.rdchem.Mol object at 0x13580f4c0>
-9 <rdkit.Chem.rdchem.Mol object at 0x13580f060>
-10 <rdkit.Chem.rdchem.Mol object at 0x13580c200>
-11 <rdkit.Chem.rdchem.Mol object at 0x13580d770>
-12 <rdkit.Chem.rdchem.Mol object at 0x13580de70>
-13 <rdkit.Chem.rdchem.Mol object at 0x13580e2d0>
-Name: mol, dtype: object
-
-
-
At this stage, nothing really clicked at the moment, but then I thought about how Datamol was built on top of RDKit and also how a few other cheminformatics posts I’ve read before utilised the functions in RDKit, so it was time to look deeper in RDKit to search for methods with the intended purpose in mind. I then found the SmilesWriter() method from RDKit after reading a few online references.
-
I’ve found out that:
-
-
To write multiple SMILES into a .smi file, use SmilesWriter()
-
To read a set of SMILES from a .smi file, use SmilesMolSupplier()
-
-
Acknowledgement of a useful link I’ve found online which had helped me to figure out how to save multiple SMILES strings in a .smi file.
-
-
# Figured out how to save multiple SMILES as a text file
-cpd3 = SmilesWriter('cpd3.smi')
-
-# Note: saving multiple SMILES strings from RDKit mol objects (cpd3_mols)
-for s in cpd3_mols:
- cpd3.write(s)
-cpd3.close()
-
-
-
-
Function for saving multiple SMILES strings as a .smi file
-
So based on the ideas in the previous section, I came up with the following simple function to save multiple SMILES strings as a .smi file.
-
-
def save_smiles_strings(df, file_name):
-
-# Create a RDKit mol column in the dataframe
- df["mol"] = df.Scaffold.apply(Chem.MolFromSmiles)
-
-# Save the "mol" column with target scaffolds as an object
- smiles_mols = df["mol"]
-
-# Use RDKit's SmilesWriter() to write the smiles strings from mol objects
-# Specify file name for .smi file, which will be stored in the working directory
- smiles = SmilesWriter(f"{file_name}.smi")
-
-# Use a for loop to iterate through each SMILES string in the dataframe
-for s in smiles_mols:
- smiles.write(s)
- smiles.close()
-
-
-
-
Testing on the function
-
Here I used one of the dataframes I’ve saved earlier, cefe_scaffolds, to test this function on saving multiple SMILES into a file. Since cefe_scaffolds dataframe was a Polars dataframe from earlier, it needed to be converted into a Pandas dataframe in order to be compatible with RDKit, which was used in the function.
-
-
# Convert Polars dataframe into a Pandas one
-cefe_scaffolds = cefe_scaffolds.to_pandas()
-
-# Running function on cefe_scaffolds dataframe
-# First parameter - dataframe to be used
-# Second parameter - file name for the SMILES strings saved
-save_smiles_strings(cefe_scaffolds, "cefe")
-
-
A .smi file with the name “cefe.smi” should appear in the working directory after running the function.
-
Now, the next stage would be to parse these SMILES strings and save them as a list. I actually worked backwards here where I looked into the Pandas.query() method first, and looked into options I could have on reading and checking for matches in multiple strings. To be able to read multiple strings in one go, a list would be suitable to carry out the matching queries (note: in the scaffold_finder library, this dataframe query method was also used in its “find_scaffolds” and “get_molecules_with_scaffold” functions).
-
An example of Pandas.query() tests:
-
-
# Using the test dataset from earlier - list of top two scaffolds with highest frequency of occurrences from ChEMBL dataset
-count_top_scaffold
# To demonstrate that querying the two top scaffolds
-# will bring back all the anti-bacterials with the same scaffold
-match_df = mol_df.query("Scaffold in @count_top_scaffold")
-match_df
-
-
-
-
-
-
-
-
-
Scaffold
-
NumAtoms
-
NumRgroupgs
-
Name
-
SMILES
-
-
-
-
-
102
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
30
-
1
-
sarecycline
-
CON(C)Cc1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C...
-
-
-
128
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
-
29
-
2
-
sarecycline
-
CON(C)Cc1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C...
-
-
-
92
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
-
29
-
3
-
tigecycline
-
CN(C)c1cc(NC(=O)CNC(C)(C)C)c(O)c2c1C[C@H]1C[C@...
-
-
-
115
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
-
29
-
3
-
demeclocycline
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
-
-
129
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
30
-
2
-
demeclocycline
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
-
-
39
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
30
-
1
-
minocycline
-
CN(C)c1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C(O...
-
-
-
65
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
-
29
-
2
-
minocycline
-
CN(C)c1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C(O...
-
-
-
115
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
-
29
-
3
-
tetracycline
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
-
-
129
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
30
-
2
-
tetracycline
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
-
-
171
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
-
29
-
3
-
eravacycline
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
-
-
201
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
30
-
2
-
eravacycline
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
-
-
172
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
30
-
3
-
oxytetracycline
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
-
-
-
92
-
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
-
29
-
3
-
omadacycline
-
CN(C)c1cc(CNCC(C)(C)C)c(O)c2c1C[C@H]1C[C@H]3[C...
-
-
-
-
-
-
-
Here, all tetracycline antibiotics were brought up in the resultant dataframe.
-
As an aside from what I wanted to do, I also learnt a small trick about how to get the number of atoms in the file with multiple SMILES strings.
-
-
# Sample use of SmilesMolSupplier & GetNumAtoms()
-suppl = SmilesMolSupplier('cpd3.smi')
-
-nMols =len(suppl)
-
-for i inrange(nMols):
-
- a = suppl[i].GetNumAtoms()
-print(a)
Now, back to where I was meant to continue working, I wanted to convert these SMILES strings into RDKit molecules first.
-
-
# Reading cpd3.smi SMILES strings in text file as RDKit mol objects
-suppl = SmilesMolSupplier("cpd3.smi")
-suppl
-
-
<rdkit.Chem.rdmolfiles.SmilesMolSupplier at 0x134bf5c10>
-
-
-
This was followed by converting the “mol” objects into SMILES strings, so that we could save each SMILES string into a list.
-
-
# Initialise an empty list
-list= []
-
-for mol in suppl:
-# Convert RDKit mol objects into SMILES strings
- m = Chem.MolToSmiles(mol)
-# Add each SMILES read from filename.smi into the empty list
-list.append(m)
-
-list
Function for converting .smi file into a list to query and match scaffolds of interests
-
I then came up with the next function that would feed multiple scaffolds into a Pandas.query() to match the strings, meaning we could compare the scaffolds with each other in a dataframe.
-
-
def query_scaffolds_via_smiles(filename):
-
-# Initialise an empty list
-list= []
-# Use SmilesMolSupplier() from RDKit to read in the SMILES strings stored in .smi file
- suppl = SmilesMolSupplier(filename)
-# Use a for loop to iterate through the SMILES strings
-for mol in suppl:
-# Convert RDKit mol objects into SMILES strings
- m = Chem.MolToSmiles(mol)
-# Add each SMILES read from filename.smi into the empty list
-list.append(m)
-# Compare the SMILES with the scaffold column in dataframe
- scaffold_match_df = mol_df.query("Scaffold in @list")
-
-return scaffold_match_df
-
-
-
-
Testing on the function
-
Below was a test for this query_scaffolds_via_smiles() function using the previously made “cpd3.smi” file.
-
To show that compound 3 scaffolds literally only existed in compound 3 and not in any other prescription-only anti-bacterials based on the ChEMBL-extracted anti-infective dataset only (note: other sources not checked at this stage).
-
-
# Testing query_scaffolds_via_smiles() function
-query_scaffolds_via_smiles("cpd3.smi")
-
-
-
-
-
-
-
-
-
Scaffold
-
NumAtoms
-
NumRgroupgs
-
Name
-
SMILES
-
-
-
-
-
0
-
O=CC1CCCC(Cc2ccc(O)cc2)C1=O
-
17
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
4
-
O=CC1CCCC(Cc2ccccc2)C1=O
-
16
-
2
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
5
-
CC(=O)C1CCCC(Cc2ccc(O)cc2)C1=O
-
18
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
8
-
CC(=O)C1CCCC(Cc2ccccc2)C1=O
-
17
-
2
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
9
-
CCC(=O)C1CCCC(Cc2ccc(O)cc2)C1=O
-
19
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
11
-
CCC(=O)C1CCCC(Cc2ccccc2)C1=O
-
18
-
2
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
12
-
O=C(CCc1ccccc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
25
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
13
-
O=C1CCCCC1C(=O)CCc1ccccc1
-
17
-
2
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
14
-
CC1CCCC(C(=O)CCc2ccccc2)C1=O
-
18
-
2
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
15
-
O=C(CCc1ccccc1)C1CCCC(Cc2ccccc2)C1=O
-
24
-
2
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
16
-
O=C1CCCCC1C(=O)CCc1ccc(O)cc1
-
18
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
19
-
CC1CCCC(C(=O)CCc2ccc(O)cc2)C1=O
-
19
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
21
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccccc2)C1=O
-
25
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
22
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
26
-
1
-
Compound_3
-
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
-
-
-
-
-
-
-
Then I also tested on “cefe.smi” created before.
-
-
# Test on cefe.smi
-query_scaffolds_via_smiles("cefe.smi")
-
-
-
-
-
-
-
-
-
Scaffold
-
NumAtoms
-
NumRgroupgs
-
Name
-
SMILES
-
-
-
-
-
0
-
C[N+]1(CC2=C(C(=O)[O-])N3C(=O)[C@@H](NC(=O)/C(...
-
31
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
1
-
C[N+]1(CC2=CN3C(=O)[C@@H](NC(=O)C(=NO)c4csc(N)...
-
28
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
2
-
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=CCS[C@H]23)cs1
-
21
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
3
-
CC1=CN2C(=O)[C@@H](NC(=O)C(=NO)c3csc(N)n3)[C@H...
-
22
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
4
-
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=C(C[NH+]4CCC...
-
27
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
5
-
C[N+]1(CC2=CN3C(=O)[C@@H](NC(=O)C=NO)[C@H]3SC2...
-
22
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
6
-
C[N+]1(CC2=CN3C(=O)[C@@H](NC(=O)C(=NO)c4cscn4)...
-
27
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
7
-
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C(C(=O)[O-])=C...
-
24
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
8
-
O=C([O-])C1=CCS[C@@H]2[C@H](NC(=O)C(=NO)c3cscn...
-
23
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
9
-
CC1=C(C(=O)[O-])N2C(=O)[C@@H](NC(=O)C(=NO)c3cs...
-
25
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
10
-
CC1=C(C(=O)[O-])N2C(=O)[C@@H](NC(=O)C=NO)[C@H]...
-
19
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
11
-
CC1=C(C(=O)[O-])N2C(=O)[C@@H](NC(=O)C(=NO)c3cs...
-
24
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
12
-
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C(C(=O)[O-])=C...
-
30
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
13
-
O=C(C=NO)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[NH+]3...
-
24
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
14
-
O=C([O-])C1=C(C[NH+]2CCCC2)CS[C@@H]2[C@H](NC(=...
-
29
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
15
-
C[N+]1(CC2=C(C(=O)[O-])N3C(=O)[C@@H](NC(=O)C=N...
-
25
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
16
-
C[N+]1(CC2=C(C(=O)[O-])N3C(=O)[C@@H](NC(=O)C(=...
-
30
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
17
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C=C(C[N+]3(C)CCCC3...
-
29
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
18
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=CCS[C@H]12)c1csc(N)n1
-
22
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
19
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=CCS[C@H]12)c1cscn1
-
21
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
20
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cs...
-
23
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
21
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cscn1
-
22
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
22
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C[NH+]3CCCC3)CS[...
-
28
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
23
-
CON=CC(=O)N[C@@H]1C(=O)N2C=C(C[NH+]3CCCC3)CS[C...
-
22
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
24
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C[NH+]3CCCC3)CS[...
-
27
-
3
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
25
-
CON=CC(=O)N[C@@H]1C(=O)N2C=C(C[N+]3(C)CCCC3)CS...
-
23
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
26
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C[N+]3(C)CCCC3)C...
-
28
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
27
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=CCS[C...
-
25
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
28
-
CON=C(C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=CCS[C@H...
-
25
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
30
-
CON=C(C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=CCS[C@H...
-
24
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
31
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C)C...
-
26
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
32
-
CON=C(C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C)CS[...
-
26
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
33
-
CON=CC(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C)CS[C...
-
20
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
34
-
CON=C(C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C)CS[...
-
25
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
35
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
31
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
36
-
CON=CC(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[NH+]...
-
25
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
37
-
CON=C(C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[NH+...
-
30
-
2
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
38
-
CO/N=C\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N+...
-
26
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
39
-
CON=CC(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N+]3...
-
26
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
40
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
31
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
41
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
32
-
1
-
cefepime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
-
-
-
2
-
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=CCS[C@H]23)cs1
-
21
-
3
-
cefixime
-
C=CC1=C(C(=O)O)N2C(=O)[C@@H](NC(=O)/C(=N\OCC(=...
-
-
-
3
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=CCS[C@H]12)c1csc(N)n1
-
22
-
3
-
cefixime
-
C=CC1=C(C(=O)O)N2C(=O)[C@@H](NC(=O)/C(=N\OCC(=...
-
-
-
2
-
CC1=CN2C(=O)[C@@H](NC(=O)C(=NO)c3csc(N)n3)[C@H...
-
22
-
3
-
ceftriaxone
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(CSc3nc...
-
-
-
24
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cs...
-
23
-
2
-
ceftriaxone
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(CSc3nc...
-
-
-
25
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cscn1
-
22
-
3
-
ceftriaxone
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(CSc3nc...
-
-
-
1
-
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=CCS[C@H]23)cs1
-
21
-
2
-
cefdinir
-
C=CC1=C(C(=O)O)N2C(=O)[C@@H](NC(=O)/C(=N\O)c3c...
-
-
-
2
-
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=CCS[C@H]23)cs1
-
21
-
3
-
cefotaxime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
-
-
-
3
-
CC1=CN2C(=O)[C@@H](NC(=O)C(=NO)c3csc(N)n3)[C@H...
-
22
-
3
-
cefotaxime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
-
-
-
19
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=CCS[C@H]12)c1csc(N)n1
-
22
-
2
-
cefotaxime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
-
-
-
20
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=CCS[C@H]12)c1cscn1
-
21
-
3
-
cefotaxime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
-
-
-
21
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cs...
-
23
-
2
-
cefotaxime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
-
-
-
22
-
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cscn1
-
22
-
3
-
cefotaxime
-
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
-
-
-
33
-
CC1=CN2C(=O)[C@@H](NC(=O)C(=NO)c3csc(N)n3)[C@H...
-
22
-
3
-
ceftazidime
-
CC(C)(O/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=...
-
-
-
36
-
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C(C(=O)[O-])=C...
-
24
-
2
-
ceftazidime
-
CC(C)(O/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=...
-
-
-
37
-
O=C([O-])C1=CCS[C@@H]2[C@H](NC(=O)C(=NO)c3cscn...
-
23
-
3
-
ceftazidime
-
CC(C)(O/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=...
-
-
-
38
-
CC1=C(C(=O)[O-])N2C(=O)[C@@H](NC(=O)C(=NO)c3cs...
-
25
-
2
-
ceftazidime
-
CC(C)(O/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=...
-
-
-
39
-
CC1=C(C(=O)[O-])N2C(=O)[C@@H](NC(=O)C(=NO)c3cs...
-
24
-
3
-
ceftazidime
-
CC(C)(O/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=...
-
-
-
-
-
-
-
Only cephalosporins were brought up as the result from this scaffold query, which matched with the antibiotic class of cefepime.
-
-
-
-
-
-
Some answers
-
There were no other prescription-only anti-bacterials from ChEMBL database with the same scaffold as compound 3 after comparing the SMILES strings between these selected scaffolds only. This was only limited to the dataset obtained from ChEMBL at this stage, with compound indications limited to anti-infectives for now. This might imply that the scaffold of compound 3 could be considered novel when comparing with molecules with similar indications. Obviously, this was too preliminary to confirm anything substantial, since there were no in vivo tests on finding out the efficacy, safety and toxicity of compound 3 apart from the in vitro experimental results mentioned in the paper. It probably would provide some ideas for scaffold hopping in hit compounds, or functional group (R-group) comparisons when looking for new compounds for synthesis. Overall, it was interesting to have a re-visit on this work using a more cheminformatics approach.
-
-
-
-
Afterthoughts
-
What I wanted to achieve in this post were:
-
-
To familiarise myself with Datamol and scaffold_finder libraries
-
To use Polars dataframe library for initial data wrangling along with Datamol Python library, and the later trial of scaffold_finder library. Using Polars would be a small degree only, as Datamol was likely written with Pandas in mind mostly (based on RDKit), while Pandas was also the more commonly used dataframe libray in many cheminformatics packages. Some people might prefer to stick with Pandas all the way, which I agree, but I’d just wanted to use Polars for the initial data wrangling only as I’ve been using it more lately
-
To reveal my thought process on building simple cheminformatics-related functions (this was unplanned, but kind of evolved while working on this post)
-
To show some evidence of my own growth from computational and medicinal chemistry with no code, to using data science tools with Python code to help guiding drug discovery projects
-
To mention that experimental validations will always be crucial for computational predictions, and since I had some experimental results from the paper, I thought to accompany it with some computational findings here
-
-
I hope I’ve at least achieved some these points in this post if not all.
-
Thanks for reading and looking forward to comments if any.
-
-
-
-
-
-
Footnotes
-
-
-
Simplified Molecular Input Line Entry Systems↩︎
-
SELF-referencIng Embedded Strings↩︎
-
International Chemical Identifier↩︎
-
-
]]>
- RDKit
- Datamol
- Python
- Pandas
- Polars
- Cheminformatics
- https://jhylin.github.io/Data_in_life_blog/posts/14_Scaffolds_in_small_molecules/chembl_anti-inf_data_prep_current.html
- Wed, 05 Jul 2023 12:00:00 GMT
-
-
- Shinylive app in Python
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_app_embed_pyodide_http.html
-
-
Quick update
-
I’ve changed the way of importing a local text/csv file from manually copying-and-pasting to using pyodide.http.open_url() in the shinylive app, which works great and avoids the clumsy manual file input. I couldn’t quite grasp the code back then, but managed to get it this time when I re-visited the problem, while also figured out that I could use the raw content link for the file from my GitHub repository. This method was also inspired by the same user answering the query in this GitHub discussion. So I’ve basically trialled both ways as suggested, which have all worked.
-
Note: if importing binary files, use pyodide.http.pyfetch() instead - check out Pyodide for details and latest changes.
-
-
-
Shinylive app in action
-
Note: it may take a few minutes to load the app (code provided at the top, with app at the bottom).
-
-
#| standalone: true
-#| components: [editor, viewer]
-#| layout: vertical
-#| viewerHeight: 420
-
-## file: app.py
-# ***Import all libraries or packages needed***
-# Import shiny ui, app
-from shiny import ui, App
-# Import shinywidgets
-from shinywidgets import output_widget, render_widget
-# Import shinyswatch to add themes
-#import shinyswatch
-# Import plotly express
-import plotly.express as px
-# Import pandas
-import pandas as pd
-# Import pyodide http - for importing file via URL
-import pyodide.http
-from pyodide.http import open_url
-
-
-# ***Specify data source***
-# Using pyodide.http.open_url
-df = pd.read_csv(open_url('https://raw.githubusercontent.com/jhylin/Data_in_life_blog/main/posts/13_Shiny_app_python/pc_cov_pd.csv'))
-
-
-# User interface---
-# Add inputs & outputs
-app_ui = ui.page_fluid(
- # Add theme - seems to only work in VS code and shinyapps.io
- #shinyswatch.theme.superhero(),
- # Add heading
- ui.h3("Molecular properties of compounds used in COVID-19 clinical trials"),
- # Place selection boxes & texts in same row
- ui.row(
- # Divide the row into two columns
- # Column 1 - selection drop-down boxes x 2
- ui.column(
- 4, ui.input_select(
- # Specify x variable input
- "x", label = "x axis:",
- choices = ["Partition coefficients",
- "Complexity",
- "Heavy atom count",
- "Hydrogen bond donor count",
- "Hydrogen bond acceptor count",
- "Rotatable bond count",
- "Molecular weight",
- "Exact mass",
- "Polar surface area",
- "Total atom stereocenter count",
- "Total bond stereocenter count"],
- ),
- ui.input_select(
- # Specify y variable input
- "y", label = "y axis:",
- choices = ["Partition coefficients",
- "Complexity",
- "Heavy atom count",
- "Hydrogen bond donor count",
- "Hydrogen bond acceptor count",
- "Rotatable bond count",
- "Molecular weight",
- "Exact mass",
- "Polar surface area",
- "Total atom stereocenter count",
- "Total bond stereocenter count"]
- )),
- # Column 2 - add texts regarding plots
- ui.column(
- 8,
- ui.p("Select different molecular properties as x and y axes to produce a scatter plot."),
- ui.tags.ul(
- ui.tags.li(
- """
- Part_coef_group means groups of partition coefficient (xlogp) as shown in the legend on the right"""
- ),
- ui.tags.li(
- """
- Toggle each partition coefficient category by clicking on the group names"""
- ),
- ui.tags.li(
- """
- Hover over each data point to see compound name and relevant molecular properties"""
- )
- )),
- # Output as a widget (interactive plot)
- output_widget("my_widget"),
- # Add texts for data source
- ui.row(
- ui.p(
- """
- Data curated by PubChem, accessed from: https://pubchem.ncbi.nlm.nih.gov/#tab=compound&query=covid-19%20clinicaltrials (last access date: 30th Apr 2023)"""
- )
- )
- )
-)
-
-
-# Server---
-# Add plotting code within my_widget function within the server function
-def server(input, output, session):
- @output
- @render_widget
- def my_widget():
- fig = px.scatter(
- df, x = input.x(), y = input.y(),
- color = "Part_coef_group",
- hover_name = "Compound name"
- )
- fig.layout.height = 400
- return fig
-
-# Combine UI & server into Shiny app
-app = App(app_ui, server)
-
-
-
-
-
-
- ]]>
- Python
- Shiny
- Pandas
- Plotly
- PubChem
- Cheminformatics
- https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_app_embed_pyodide_http.html
- Tue, 23 May 2023 12:00:00 GMT
-
-
- Shinylive app in Python
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_app_embed.html
-
-
Coding for Shinylive app in Python
-
The entire code for this Shiny app in Python were written in VS Code initially. When I was looking for places to deploy the app, I then migrated the code to RStudio IDE (note: recently I saw Python code in Quarto documents being used in VS Code after I’ve deployed this app, so this might be another option).
-
For data preparation stage, please visit this post for details.
-
-
-
Trialling open-source large-language models
-
There have been a lot of hypes surrounding large-language models (LLMs) or generative pre-trained transformers (GPTs). I’m still somewhat both reserved and excited about them at present. The area I’m most concerned with is where are the relevant laws, regulations and ethics on using these tools in public domains, whether for private or commercial uses, and will they be country-specific or globally streamlined? While I have these questions in mind, I’m not denying the fact that they can be useful for a few well-known purposes in coding, such as requesting for regex code templates for text cleaning or asking for code snippets while programmers are busy with several tasks at the same time.
-
I thought to trial out the open-source LLMs a little bit when I was working on this app, since they were the open and transparent versions with potentials for personal or commercial uses. So I’ve tested about 3 different open-source LLMs, which were H2OGPT, HuggingChat and StableLM. I decided to only give minimal amount of prompts (I’ve given H2OGPT three prompts, and only one prompt each for either HuggingChat and StableLM), and the question was framed around providing a code outline for building a Shiny app using Python and Polars dataframe libary, and see what answers they could each provide.
-
All of them produced answers that were close to what I’ve asked for, i.e. I could see a code outline for a Shiny app.py file at first but it was more like R code, with one case mixing both Python and R in the code concurrently. However, none of them actually reached exactly how I’ve asked them to provide, perhaps if I’ve given more prompts then they might manage to provide answers closer to the request. Also, none of them were able to pick up the more recent Polars dataframe library (which were probably more prominent from 2021 onwards) and also Shiny in Python (which was also quite new as well, only out of alpha phase recently). All of them only showed code using Pandas dataframe libray, even though I did mention Polars in all of the question prompts. Once I get to use more of these open-source LLMs in the future, which currently are gaining more traction from what I’ve read online, I think I might try to write another post about them later.
-
-
-
-
Using Shiny in Python documentations
-
After trialling open-source LLMs a bit to help with the coding part (although not as helpful as I first imagined, but they still somewhat provided a rough code framework for Shiny apps), I completed the app.py script in the end by using mainly documents from Shiny for Python website. I still think thorough documentations for any products are still fundamental and inevitable, because these documentations will be sources to provide high-quality data for training these LLMs which means they’ll be of higher quality too.
-
Several links I’ve used and found to be very informative:
-
-
“Shiny for Python out of alpha” - quick summary on recent status update and new features such as shinyswatch and shinywidgets on Shiny for Python
-
“Quickstart for R users” - a very useful link for people who are R users or already familiar with Shiny apps in R, and would like to build Shiny apps in Python
After building the app in a workable condition, I started looking at where and how to deploy this Shiny app written in Python. Initially I used the easiest method as stated from the Shinylive link above, which was to deploy on Shinylive editor. One drawback for this was that the application URL was extremely long as the URL hash would store the entire app code inside. However, this was indeed one of the simplest ways to share the apps with others quickly, by simply providing the URL link with the intended audience.
-
I then went on to try another method where I spent one afternoon trying to figure out that I had to add an install_certificate.command for my operating system since I installed Python by using Homebrew in the first place (which some clever users might already know to avoid…), so that I could resolve the SSL certificate issue if I’m trying to connect with Shinyapps.io.
-
I also tried to follow another method from an earlier time which suggested to deploy the app directly on GitHub Pages, but since then the code was changed and updated to a different version and the old method no longer worked as nicely as mentioned (a lot of the code for Shiny in Python can still be experimental, and can change drastically, so check the sources to see the latest updates). I then stumbled upon another GitHub repository from the Quarto team at Posit/RStudio and looked into the possibility and tested it. Eventually, I settled on this method - embedding the app in a Quarto document, which provided both code and app itself on this very same webpage down below.
-
One of the downsides of the following code that I couldn’t quite get rid of yet was the poor file importation style. for loading a .csv file locally. I’ve added code annotations as comments to explain what I’ve done. The solution to import .csv file into app.py for the embedding version was from this discussion in the repository (thanks to that particular user who found this hacky way to do it). However, the plus side was that coding for the rest of the app was not bad at all and worked seamlessly. I’ll try to follow up later on whether importing .csv file for app embedding could be easier, perhaps this might be something the Quarto team is working right now.
-
The second downside was that shinyswatch package might not be functional yet for this embedding method (but most likely would work if deploying apps to shinyapps.io). This meant there would be no background visual themes yet for apps embedded in Quarto docs, but hopefully this would be possible in the future. Overall, I was amazed at how easy it was to build, deploy and embed a Shiny app in Python in a Quarto document.
-
Note: if using Shinylive editor, make sure to include a requirement.txt file if using extra Python packages such as Pandas or Plotly. For embedding apps in Quarto docs, this appears not to be compulsory and only optional when I tried it down below.
-
Code for this post (.qmd document) is available here.
-
-
-
Shinylive app in action
-
Note: it may take a few minutes to load the app (code provided at the top, with app at the bottom).
-
Update on 24/5/23: to avoid manual file input, see this updated post which uses pyodide.http.open_url() method (code change in editor section).
-
-
#| standalone: true
-#| components: [editor, viewer]
-#| layout: vertical
-#| viewerHeight: 420
-
-## file: app.py
-# ***Import all libraries or packages needed***
-# Import shiny ui, app
-from shiny import ui, App
-# Import shinywidgets
-from shinywidgets import output_widget, render_widget
-# Import shinyswatch to add themes
-#import shinyswatch
-# Import plotly express
-import plotly.express as px
-# Import pandas
-import pandas as pd
-from pathlib import Path
-
-
-# User interface---
-# Add inputs & outputs
-app_ui = ui.page_fluid(
- # Add theme - seems to only work in VS code and shinyapps.io
- #shinyswatch.theme.superhero(),
- # Add heading
- ui.h3("Molecular properties of compounds used in COVID-19 clinical trials"),
- # Place selection boxes & texts in same row
- ui.row(
- # Divide the row into two columns
- # Column 1 - selection drop-down boxes x 2
- ui.column(
- 4, ui.input_select(
- # Specify x variable input
- "x", label = "x axis:",
- choices = ["Partition coefficients",
- "Complexity",
- "Heavy atom count",
- "Hydrogen bond donor count",
- "Hydrogen bond acceptor count",
- "Rotatable bond count",
- "Molecular weight",
- "Exact mass",
- "Polar surface area",
- "Total atom stereocenter count",
- "Total bond stereocenter count"],
- ),
- ui.input_select(
- # Specify y variable input
- "y", label = "y axis:",
- choices = ["Partition coefficients",
- "Complexity",
- "Heavy atom count",
- "Hydrogen bond donor count",
- "Hydrogen bond acceptor count",
- "Rotatable bond count",
- "Molecular weight",
- "Exact mass",
- "Polar surface area",
- "Total atom stereocenter count",
- "Total bond stereocenter count"]
- )),
- # Column 2 - add texts regarding plots
- ui.column(
- 8,
- ui.p("Select different molecular properties as x and y axes to produce a scatter plot."),
- ui.tags.ul(
- ui.tags.li(
- """
- Part_coef_group means groups of partition coefficient (xlogp) as shown in the legend on the right"""
- ),
- ui.tags.li(
- """
- Toggle each partition coefficient category by clicking on the group names"""
- ),
- ui.tags.li(
- """
- Hover over each data point to see compound name and relevant molecular properties"""
- )
- )),
- # Output as a widget (interactive plot)
- output_widget("my_widget"),
- # Add texts for data source
- ui.row(
- ui.p(
- """
- Data curated by PubChem, accessed from: https://pubchem.ncbi.nlm.nih.gov/#tab=compound&query=covid-19%20clinicaltrials (last access date: 30th Apr 2023)"""
- )
- )
- )
-)
-
-
-# Server---
-# Add plotting code within my_widget function within the server function
-def server(input, output, session):
- @output
- @render_widget
- def my_widget():
- fig = px.scatter(
- df, x = input.x(), y = input.y(),
- color = "Part_coef_group",
- hover_name = "Compound name"
- )
- fig.layout.height = 400
- return fig
-
-# Combine UI & server into Shiny app
-app = App(app_ui, server)
-
-
-# ***Specify data source***
-# --Not the best approach yet but works for now--
-# Currently this work-around only suits small to medium-size dataset
-# Not ideal for large dataset for sure
-# To load file locally use the following code
-infile = Path(__file__).parent / "pc_cov_pd.csv"
-df = pd.read_csv(infile)
-
-# Then manually paste in csv/txt file below
-## file: pc_cov_pd.csv
-,Compound name,Molecular weight,Polar surface area,Complexity,Partition coefficients,Heavy atom count,Hydrogen bond donor count,Hydrogen bond acceptor count,Rotatable bond count,Exact mass,Monoisotopic mass,Formal charge,Covalently-bonded unit count,Isotope atom count,Total atom stereocenter count,Defined atom stereocenter count,Undefined atoms stereocenter count,Total bond stereocenter count,Defined bond stereocenter count,Undefined bond stereocenter count,Part_coef_group
-0,Calcitriol,416.6,60.7,688.0,5.1,30,3,3,6,416.329,416.329,0,1,0,6,6,0,2,2,0,Larger than 6
-1,Ubiquinol,865.4,58.9,1600.0,20.2,63,2,4,31,864.7,864.7,0,1,0,0,0,0,9,9,0,Larger than 6
-2,Glutamine,146.14,106.0,146.0,-3.1,10,3,4,4,146.069,146.069,0,1,0,1,1,0,0,0,0,Between -11 and 5
-3,Aspirin,180.16,63.6,212.0,1.2,13,1,4,3,180.042,180.042,0,1,0,0,0,0,0,0,0,Between -11 and 5
-4,1-Methylnicotinamide,137.16,47.0,136.0,-0.1,10,1,1,1,137.071,137.071,1,1,0,0,0,0,0,0,0,Between -11 and 5
-5,Losartan,422.9,92.5,520.0,4.3,30,2,5,8,422.162,422.162,0,1,0,0,0,0,0,0,0,Between -11 and 5
-6,Vitamin E,430.7,29.5,503.0,10.7,31,1,2,12,430.381,430.381,0,1,0,3,3,0,0,0,0,Larger than 6
-7,Nicotinamide,122.12,56.0,114.0,-0.4,9,1,2,1,122.048,122.048,0,1,0,0,0,0,0,0,0,Between -11 and 5
-8,Adenosine,267.24,140.0,335.0,-1.1,19,4,8,2,267.097,267.097,0,1,0,4,4,0,0,0,0,Between -11 and 5
-9,Inosine,268.23,129.0,405.0,-1.3,19,4,7,2,268.081,268.081,0,1,0,4,4,0,0,0,0,Between -11 and 5
-10,Nicotinamide riboside,255.25,117.0,314.0,-1.8,18,4,5,3,255.098,255.098,1,1,0,4,4,0,0,0,0,Between -11 and 5
-11,Dimethyl Fumarate,144.12,52.6,141.0,0.7,10,0,4,4,144.042,144.042,0,1,0,0,0,0,1,1,0,Between -11 and 5
-12,Inosinic acid,348.21,176.0,555.0,-3.0,23,5,10,4,348.047,348.047,0,1,0,4,4,0,0,0,0,Between -11 and 5
-13,Acetyl-L-carnitine,203.24,66.4,214.0,0.4,14,0,4,5,203.116,203.116,0,1,0,1,1,0,0,0,0,Between -11 and 5
-14,Camostat,398.4,137.0,602.0,1.1,29,2,6,9,398.159,398.159,0,1,0,0,0,0,0,0,0,Between -11 and 5
-15,Estradiol,272.4,40.5,382.0,4.0,20,2,2,0,272.178,272.178,0,1,0,5,5,0,0,0,0,Between -11 and 5
-16,Aspartic Acid,133.1,101.0,133.0,-2.8,9,3,5,3,133.038,133.038,0,1,0,1,1,0,0,0,0,Between -11 and 5
-17,Ribavirin,244.2,144.0,304.0,-1.8,17,4,7,3,244.081,244.081,0,1,0,4,4,0,0,0,0,Between -11 and 5
-18,Angiotensin (1-7),899.0,380.0,1660.0,-3.0,64,12,14,25,898.466,898.466,0,1,0,8,8,0,0,0,0,Between -11 and 5
-19,alpha-Maltose,342.3,190.0,382.0,-4.7,23,8,11,4,342.116,342.116,0,1,0,10,10,0,0,0,0,Between -11 and 5
-20,Ambrisentan,378.4,81.5,475.0,3.8,28,1,6,7,378.158,378.158,0,1,0,1,1,0,0,0,0,Between -11 and 5
-21,Ciclosporin,1202.6,279.0,2330.0,7.5,85,5,12,15,1201.84,1201.84,0,1,0,12,12,0,1,1,0,Larger than 6
-22,Ergocalciferol,396.6,20.2,678.0,7.4,29,1,1,5,396.339,396.339,0,1,0,6,6,0,3,3,0,Larger than 6
-23,Docosahexaenoic Acid,328.5,37.3,462.0,6.2,24,1,2,14,328.24,328.24,0,1,0,0,0,0,6,6,0,Larger than 6
-24,Tacrolimus,804.0,178.0,1480.0,2.7,57,3,12,7,803.482,803.482,0,1,0,14,14,0,2,2,0,Between -11 and 5
-25,Budesonide,430.5,93.1,862.0,2.5,31,2,6,4,430.236,430.236,0,1,0,9,8,1,0,0,0,Between -11 and 5
-26,Calcifediol,400.6,40.5,655.0,6.2,29,2,2,6,400.334,400.334,0,1,0,5,5,0,2,2,0,Larger than 6
-27,Cepharanthine,606.7,61.9,994.0,6.5,45,0,8,2,606.273,606.273,0,1,0,2,2,0,0,0,0,Larger than 6
-28,Cholecalciferol,384.6,20.2,610.0,7.9,28,1,1,6,384.339,384.339,0,1,0,5,5,0,2,2,0,Larger than 6
-29,Coenzyme Q10,863.3,52.6,1840.0,19.4,63,0,4,31,862.684,862.684,0,1,0,0,0,0,9,9,0,Larger than 6
-30,Tacrolimus monohydrate,822.0,179.0,1480.0,,58,4,13,7,821.493,821.493,0,2,0,14,14,0,2,2,0,Larger than 6
-31,Alisporivir,1216.6,279.0,2360.0,7.9,86,5,12,15,1215.86,1215.86,0,1,0,13,13,0,1,1,0,Larger than 6
-32,Pacritinib,472.6,68.7,644.0,3.8,35,1,7,4,472.247,472.247,0,1,0,0,0,0,1,1,0,Between -11 and 5
-33,Topotecan,421.4,103.0,867.0,0.5,31,2,7,3,421.164,421.164,0,1,0,1,1,0,0,0,0,Between -11 and 5
-34,Voclosporin,1214.6,279.0,2380.0,7.9,86,5,12,16,1213.84,1213.84,0,1,0,12,12,0,1,1,0,Larger than 6
-35,"(1R,4S,5'S,6R,6'R,8R,10E,12S,13S,14E,16E,20R,21R,24S)-6'-[(2R)-Butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,3,0,Between -11 and 5
-36,Oxytocin,1007.2,450.0,1870.0,-2.6,69,12,15,17,1006.44,1006.44,0,1,0,9,9,0,0,0,0,Between -11 and 5
-37,"(1R,4S,6R,10E,14E,16E,21R)-6'-butan-2-yl-21,24-dihydroxy-12-[(2R,4S,6S)-5-[(2S,4S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,10,10,3,3,0,Between -11 and 5
-38,"Manganese, dichloro((4aS,13aS,17aS,21aS)-1,2,3,4,4a,5,6,12,13,13a,14,15,16,17,17a,18,19,20,21,21a-eicosahydro-7,11-nitrilo-7H-dibenzo(b,H)-5,13,18,21-tetraazacycloheptadecine-kappaN5,kappaN13,kappaN18,kappaN21,kappaN22)-, (pb-7-11-2344'3')-",483.4,61.0,381.0,,29,4,7,0,482.165,482.165,0,4,0,4,4,0,0,0,0,Larger than 6
-39,Ivermectin B1a,875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,3,0,Between -11 and 5
-40,Dipyridamole,504.6,145.0,561.0,0.7,36,4,12,12,504.317,504.317,0,1,0,0,0,0,0,0,0,Between -11 and 5
-41,Tetrandrine,622.7,61.9,979.0,6.4,46,0,8,4,622.304,622.304,0,1,0,2,2,0,0,0,0,Larger than 6
-42,Sirolimus,914.2,195.0,1760.0,6.0,65,3,13,6,913.555,913.555,0,1,0,15,15,0,4,4,0,Larger than 6
-43,Iloprost,360.5,77.8,606.0,2.8,26,3,4,8,360.23,360.23,0,1,0,6,5,1,2,2,0,Between -11 and 5
-44,Ramipril,416.5,95.9,619.0,1.4,30,2,6,10,416.231,416.231,0,1,0,5,5,0,0,0,0,Between -11 and 5
-45,Prasugrel hydrochloride,409.9,74.8,555.0,,27,1,6,6,409.091,409.091,0,2,0,1,0,1,0,0,0,Larger than 6
-46,Uproleselan,1304.5,383.0,1870.0,-2.0,90,9,27,52,1303.72,1303.72,0,1,0,15,15,0,0,0,0,Between -11 and 5
-47,MET-enkephalin,573.7,225.0,847.0,-2.1,40,7,9,16,573.226,573.226,0,1,0,3,3,0,0,0,0,Between -11 and 5
-48,"(18Z)-1,14-dihydroxy-12-[(E)-1-(4-hydroxy-3-methoxycyclohexyl)prop-1-en-2-yl]-23,25-dimethoxy-13,19,21,27-tetramethyl-17-prop-2-enyl-11,28-dioxa-4-azatricyclo[22.3.1.04,9]octacos-18-ene-2,3,10,16-tetrone",804.0,178.0,1480.0,2.7,57,3,12,7,803.482,803.482,0,1,0,14,0,14,2,2,0,Between -11 and 5
-49,"(1R,4S,5'S,6R,6'R,8R,10E,13S,14E,16E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,19,1,3,3,0,Between -11 and 5
-50,Plitidepsin,1110.3,285.0,2200.0,5.7,79,4,15,15,1109.63,1109.63,0,1,0,12,12,0,0,0,0,Larger than 6
-51,"(10Z,14Z,16Z)-6'-butan-2-yl-21,24-dihydroxy-12-[5-(5-hydroxy-4-methoxy-6-methyloxan-2-yl)oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,0,20,3,3,0,Between -11 and 5
-52,"(1R,4S,5'S,6R,6'R,8R,12S,13S,20R,21R,24S)-6'-[(2R)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,0,3,Between -11 and 5
-53,Rubramin,1355.4,476.0,3220.0,,93,9,21,16,1354.57,1354.57,-3,3,0,14,14,0,3,3,0,Larger than 6
-54,"(1R,4S,5'S,6R,6'S,8R,10E,12S,13S,14E,16E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,3,0,Between -11 and 5
-55,Zidovudine,267.24,93.2,484.0,0.0,19,2,6,3,267.097,267.097,0,1,0,3,3,0,0,0,0,Between -11 and 5
-56,Resveratrol,228.24,60.7,246.0,3.1,17,3,3,2,228.079,228.079,0,1,0,0,0,0,1,1,0,Between -11 and 5
-57,Curcumin,368.4,93.1,507.0,3.2,27,2,6,8,368.126,368.126,0,1,0,0,0,0,2,2,0,Between -11 and 5
-58,"6'-Butan-2-yl-21,24-dihydroxy-12-[5-(5-hydroxy-4-methoxy-6-methyloxan-2-yl)oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,0,20,3,0,3,Between -11 and 5
-59,Regorafenib,482.8,92.4,686.0,4.2,33,3,8,5,482.077,482.077,0,1,0,0,0,0,0,0,0,Between -11 and 5
-60,Pharmakon1600-01300027,875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,17,3,3,3,0,Between -11 and 5
-61,"(1R,4S,5'S,6R,6'R,8R,10Z,12S,13S,14Z,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,2,1,Between -11 and 5
-62,"(1R,4S,5'S,6R,6'R,8R,10E,12S,13S,14E,16E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4R,5S,6S)-5-[(2S,4R,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,3,0,Between -11 and 5
-63,Methylcobalamin,1345.4,449.0,3160.0,,92,10,20,26,1344.6,1344.6,0,3,0,14,13,1,3,3,0,Larger than 6
-64,Spironolactone,416.6,85.7,818.0,2.9,29,0,5,2,416.202,416.202,0,1,0,7,7,0,0,0,0,Between -11 and 5
-65,Prednisone,358.4,91.7,764.0,1.5,26,2,5,2,358.178,358.178,0,1,0,6,6,0,0,0,0,Between -11 and 5
-66,Quercetin,302.23,127.0,488.0,1.5,22,5,7,1,302.043,302.043,0,1,0,0,0,0,0,0,0,Between -11 and 5
-67,Calderol,418.7,41.5,655.0,,30,3,3,6,418.345,418.345,0,2,0,5,5,0,2,2,0,Larger than 6
-68,Dactolisib,469.5,73.1,872.0,5.2,36,0,4,3,469.19,469.19,0,1,0,0,0,0,0,0,0,Larger than 6
-69,Enzalutamide,464.4,109.0,839.0,3.6,32,1,8,3,464.093,464.093,0,1,0,0,0,0,0,0,0,Between -11 and 5
-70,Selinexor,443.3,97.6,621.0,3.0,31,2,12,5,443.093,443.093,0,1,0,0,0,0,1,1,0,Between -11 and 5
-71,Chlorpromazine,318.9,31.8,339.0,5.2,21,0,3,4,318.096,318.096,0,1,0,0,0,0,0,0,0,Larger than 6
-72,Itraconazolum [Latin],705.6,101.0,1120.0,5.7,49,0,9,11,704.239,704.239,0,1,0,3,0,3,0,0,0,Larger than 6
-73,Loratadine,382.9,42.4,569.0,5.2,27,0,3,2,382.145,382.145,0,1,0,0,0,0,0,0,0,Larger than 6
-74,Nifedipine,346.3,110.0,608.0,2.2,25,1,7,5,346.116,346.116,0,1,0,0,0,0,0,0,0,Between -11 and 5
-75,Prazosin,383.4,107.0,544.0,2.0,28,1,8,4,383.159,383.159,0,1,0,0,0,0,0,0,0,Between -11 and 5
-76,Alizarin,240.21,74.6,378.0,3.2,18,2,4,0,240.042,240.042,0,1,0,0,0,0,0,0,0,Between -11 and 5
-77,Methylprednisolone,374.5,94.8,754.0,1.9,27,3,5,2,374.209,374.209,0,1,0,8,8,0,0,0,0,Between -11 and 5
-78,Chloroquine sulfate,418.0,111.0,390.0,,27,3,7,8,417.149,417.149,0,2,0,1,0,1,0,0,0,Larger than 6
-79,Bardoxolone methyl,505.7,84.2,1210.0,6.7,37,0,5,2,505.319,505.319,0,1,0,7,7,0,0,0,0,Larger than 6
-80,"(1R,4S,5'S,6R,6'R,8R,12S,13S,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,0,3,Between -11 and 5
-81,Heartgard-30,1736.2,340.0,3340.0,,123,6,28,15,1736.0,1735.0,0,2,0,39,34,5,6,6,0,Larger than 6
-82,Ivermectine 100 microg/mL in Acetonitrile,1736.2,340.0,3340.0,,123,6,28,15,1736.0,1735.0,0,2,0,39,39,0,6,6,0,Larger than 6
-83,Duvelisib,416.9,86.8,668.0,4.1,30,2,5,4,416.115,416.115,0,1,0,1,1,0,0,0,0,Between -11 and 5
-84,"(4S,5'S,6R,6'R,8R,10E,12S,13S,14E,16E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,19,1,3,3,0,Between -11 and 5
-85,Zotatifin,487.5,108.0,819.0,2.4,36,2,8,6,487.211,487.211,0,1,0,5,5,0,0,0,0,Between -11 and 5
-86,MaxEPA,645.0,74.6,874.0,,47,2,4,28,644.48,644.48,0,2,0,0,0,0,11,11,0,Larger than 6
-87,"(1R,4S,5'S,6R,6'R,8R,12S,13S,14E,16E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,2,1,Between -11 and 5
-88,"(1S,4S,5'S,6R,6'R,8R,10E,12S,13S,14E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,2,1,Between -11 and 5
-89,Indomethacin,357.8,68.5,506.0,4.3,25,1,4,4,357.077,357.077,0,1,0,0,0,0,0,0,0,Between -11 and 5
-90,Methylene Blue,319.9,43.9,483.0,,21,0,4,1,319.091,319.091,0,2,0,0,0,0,0,0,0,Larger than 6
-91,Bromocresol green,698.0,92.2,742.0,6.5,31,2,5,2,697.725,693.73,0,1,0,0,0,0,0,0,0,Larger than 6
-92,Hydroxychloroquine sulfate,434.0,131.0,413.0,,28,4,8,9,433.144,433.144,0,2,0,1,0,1,0,0,0,Larger than 6
-93,Ritonavir,720.9,202.0,1040.0,6.0,50,4,9,18,720.313,720.313,0,1,0,4,4,0,0,0,0,Larger than 6
-94,Fisetin,286.24,107.0,459.0,2.0,21,4,6,1,286.048,286.048,0,1,0,0,0,0,0,0,0,Between -11 and 5
-95,Palbociclib,447.5,103.0,775.0,1.8,33,2,8,5,447.238,447.238,0,1,0,0,0,0,0,0,0,Between -11 and 5
-96,Ruxolitinib,306.4,83.2,453.0,2.1,23,1,4,4,306.159,306.159,0,1,0,1,1,0,0,0,0,Between -11 and 5
-97,Sildenafil Citrate,666.7,250.0,1070.0,,46,5,15,12,666.232,666.232,0,2,0,0,0,0,0,0,0,Larger than 6
-98,Nintedanib,539.6,102.0,892.0,4.3,40,2,7,8,539.253,539.253,0,1,0,0,0,0,0,0,0,Between -11 and 5
-99,Melatonin,232.28,54.1,270.0,0.8,17,2,2,4,232.121,232.121,0,1,0,0,0,0,0,0,0,Between -11 and 5
-100,Carbamazepine,236.27,46.3,326.0,2.5,18,1,1,0,236.095,236.095,0,1,0,0,0,0,0,0,0,Between -11 and 5
-101,Clofazimine,473.4,40.0,829.0,7.1,33,1,4,4,472.122,472.122,0,1,0,0,0,0,0,0,0,Larger than 6
-102,Diphenhydramine,255.35,12.5,211.0,3.3,19,0,2,6,255.162,255.162,0,1,0,0,0,0,0,0,0,Between -11 and 5
-103,Thalidomide,258.23,83.6,449.0,0.3,19,1,4,1,258.064,258.064,0,1,0,1,0,1,0,0,0,Between -11 and 5
-104,Hydrocortisone,362.5,94.8,684.0,1.6,26,3,5,2,362.209,362.209,0,1,0,7,7,0,0,0,0,Between -11 and 5
-105,Progesterone,314.5,34.1,589.0,3.9,23,0,2,1,314.225,314.225,0,1,0,6,6,0,0,0,0,Between -11 and 5
-106,o-Aminoazotoluene,225.29,50.7,264.0,3.7,17,1,3,2,225.127,225.127,0,1,0,0,0,0,0,0,0,Between -11 and 5
-107,Clonitralid,388.2,141.0,414.0,,25,4,6,3,387.039,387.039,0,2,0,0,0,0,0,0,0,Larger than 6
-108,Pimozide,461.5,35.6,632.0,6.3,34,1,4,7,461.228,461.228,0,1,0,0,0,0,0,0,0,Larger than 6
-109,Nitazoxanide,307.28,142.0,428.0,2.0,21,1,7,4,307.026,307.026,0,1,0,0,0,0,0,0,0,Between -11 and 5
-110,Deferoxamine hydrochloride,597.1,206.0,739.0,,40,7,9,23,596.33,596.33,0,2,0,0,0,0,0,0,0,Larger than 6
-111,Chloroquine monophosphate,417.9,106.0,359.0,,27,4,7,8,417.158,417.158,0,2,0,1,0,1,0,0,0,Larger than 6
-112,Retinol,286.5,20.2,496.0,5.7,21,1,1,5,286.23,286.23,0,1,0,0,0,0,4,4,0,Larger than 6
-113,Melphalan,305.2,66.6,265.0,-0.5,19,2,4,8,304.075,304.075,0,1,0,1,1,0,0,0,0,Between -11 and 5
-114,Dasatinib,488.0,135.0,642.0,3.6,33,3,9,7,487.156,487.156,0,1,0,0,0,0,0,0,0,Between -11 and 5
-115,Masitinib,498.6,102.0,696.0,4.3,36,2,7,7,498.22,498.22,0,1,0,0,0,0,0,0,0,Between -11 and 5
-116,Acrivastine and pseudoephedrine hydrochloride,550.1,85.7,635.0,,39,4,6,9,549.276,549.276,0,3,0,2,2,0,2,2,0,Larger than 6
-117,"4-(carboxymethyl)-2-((R)-1-(2-(2,5-dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-1,3,2-dioxaborinane-4-carboxylic acid",517.1,168.0,815.0,,34,4,9,10,516.087,516.087,0,1,0,2,1,1,0,0,0,Larger than 6
-118,Remdesivir,602.6,204.0,1010.0,1.9,42,4,13,14,602.225,602.225,0,1,0,6,6,0,0,0,0,Between -11 and 5
-119,"Ivermectin B1a, epi-",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,3,0,Between -11 and 5
-120,Hydroxocobalamin,1270.4,452.0,3140.0,-3.4,90,9,19,26,1269.63,1269.63,-2,1,0,14,0,14,3,3,0,Between -11 and 5
-121,Zanubrutinib,471.5,103.0,756.0,3.5,35,2,5,6,471.227,471.227,0,1,0,1,1,0,0,0,0,Between -11 and 5
-122,Gamma-Aminobutyric Acid,103.12,63.3,62.7,-3.2,7,2,3,3,103.063,103.063,0,1,0,0,0,0,0,0,0,Between -11 and 5
-123,Caffeine,194.19,58.4,293.0,-0.1,14,0,3,0,194.08,194.08,0,1,0,0,0,0,0,0,0,Between -11 and 5
-124,Dapsone,248.3,94.6,306.0,1.0,17,2,4,2,248.062,248.062,0,1,0,0,0,0,0,0,0,Between -11 and 5
-125,Leflunomide,270.21,55.1,327.0,2.5,19,1,6,2,270.062,270.062,0,1,0,0,0,0,0,0,0,Between -11 and 5
-126,Raloxifene,473.6,98.2,655.0,6.1,34,2,6,7,473.166,473.166,0,1,0,0,0,0,0,0,0,Larger than 6
-127,Imatinib,493.6,86.3,706.0,3.5,37,2,7,7,493.259,493.259,0,1,0,0,0,0,0,0,0,Between -11 and 5
-128,Dexamethasone,392.5,94.8,805.0,1.9,28,3,6,2,392.2,392.2,0,1,0,8,8,0,0,0,0,Between -11 and 5
-129,Colchicine,399.4,83.1,740.0,1.0,29,1,6,5,399.168,399.168,0,1,0,1,1,0,0,0,0,Between -11 and 5
-130,Estradiol cypionate,396.6,46.5,597.0,7.1,29,1,3,5,396.266,396.266,0,1,0,5,5,0,0,0,0,Larger than 6
-131,Cetylpyridinium Chloride,340.0,3.9,208.0,,23,0,1,15,339.269,339.269,0,2,0,0,0,0,0,0,0,Larger than 6
-132,Plerixafor,502.8,78.7,456.0,0.0,36,6,8,4,502.447,502.447,0,1,0,0,0,0,0,0,0,Between -11 and 5
-133,Telmisartan,514.6,72.9,831.0,6.9,39,1,4,7,514.237,514.237,0,1,0,0,0,0,0,0,0,Larger than 6
-134,Sorafenib,464.8,92.4,646.0,4.1,32,3,7,5,464.086,464.086,0,1,0,0,0,0,0,0,0,Between -11 and 5
-135,Bacitracin zinc,1488.1,552.0,2950.0,,101,16,21,31,1485.68,1485.68,0,2,0,16,0,16,0,0,0,Larger than 6
-136,Hymecromone,176.17,46.5,257.0,1.9,13,1,3,0,176.047,176.047,0,1,0,0,0,0,0,0,0,Between -11 and 5
-137,Cob(II)alamin,1329.3,452.0,3150.0,,91,9,19,16,1328.56,1328.56,-2,2,0,14,14,0,3,3,0,Larger than 6
-138,Tofacitinib,312.37,88.9,488.0,1.5,23,1,5,3,312.17,312.17,0,1,0,2,2,0,0,0,0,Between -11 and 5
-139,Elsulfavirine,629.3,134.0,977.0,5.2,37,2,7,8,626.943,626.943,0,1,0,0,0,0,0,0,0,Larger than 6
-140,Fostamatinib,580.5,187.0,904.0,1.6,40,4,15,10,580.148,580.148,0,1,0,0,0,0,0,0,0,Between -11 and 5
-141,3-(4-chlorophenyl)-N-(pyridin-4-ylmethyl)adamantane-1-carboxamide,380.9,42.0,551.0,4.7,27,1,2,4,380.166,380.166,0,1,0,2,0,2,0,0,0,Between -11 and 5
-142,Adenosylcobalamin,1579.6,571.0,3730.0,,109,12,27,17,1578.66,1578.66,-3,3,0,18,18,0,3,3,0,Larger than 6
-143,Solnatide,1923.1,861.0,4170.0,-10.7,134,27,32,27,1921.81,1921.81,0,1,0,16,16,0,0,0,0,Smaller than -10
-144,Subasumstat,578.1,193.0,942.0,3.5,38,4,11,8,577.122,577.122,0,1,0,4,4,0,0,0,0,Between -11 and 5
-145,Lactoferrin,3125.8,1360.0,7330.0,6.8,219,51,65,108,3124.68,3123.68,0,1,0,26,26,0,0,0,0,Larger than 6
-146,Compstatin 40,1789.1,692.0,3770.0,-2.1,126,21,23,30,1787.84,1787.84,0,1,0,15,15,0,0,0,0,Between -11 and 5
-147,Celecoxib,381.4,86.4,577.0,3.4,26,1,7,3,381.076,381.076,0,1,0,0,0,0,0,0,0,Between -11 and 5
-148,Eucalyptol,154.25,9.2,164.0,2.5,11,0,1,0,154.136,154.136,0,1,0,0,0,0,0,0,0,Between -11 and 5
-149,Disulfiram,296.5,121.0,201.0,3.9,16,0,4,7,296.051,296.051,0,1,0,0,0,0,0,0,0,Between -11 and 5
-150,Ibuprofen,206.28,37.3,203.0,3.5,15,1,2,4,206.131,206.131,0,1,0,1,0,1,0,0,0,Between -11 and 5
-151,Niclosamide,327.12,95.2,404.0,4.0,21,2,4,2,325.986,325.986,0,1,0,0,0,0,0,0,0,Between -11 and 5
-152,Simvastatin,418.6,72.8,706.0,4.7,30,1,5,7,418.272,418.272,0,1,0,7,7,0,0,0,0,Between -11 and 5
-153,Methotrexate,454.4,211.0,704.0,-1.8,33,5,12,9,454.171,454.171,0,1,0,1,1,0,0,0,0,Between -11 and 5
-154,Angiotensin II,1046.2,409.0,1980.0,-1.7,75,13,15,29,1045.53,1045.53,0,1,0,9,9,0,0,0,0,Between -11 and 5
-155,Lenalidomide,259.26,92.5,437.0,-0.5,19,2,4,1,259.096,259.096,0,1,0,1,0,1,0,0,0,Between -11 and 5
-156,Aspartyl-alanyl-diketopiperazine,186.17,95.5,263.0,-1.4,13,3,4,2,186.064,186.064,0,1,0,2,2,0,0,0,0,Between -11 and 5
-157,Silmitasertib,349.8,75.1,491.0,4.4,25,2,5,3,349.062,349.062,0,1,0,0,0,0,0,0,0,Between -11 and 5
-158,Ibrutinib,440.5,99.2,678.0,3.6,33,1,6,5,440.196,440.196,0,1,0,1,1,0,0,0,0,Between -11 and 5
-159,Bemcentinib,506.6,97.8,775.0,5.5,38,2,7,4,506.291,506.291,0,1,0,1,1,0,0,0,0,Larger than 6
-160,Teriflunomide,270.21,73.1,426.0,3.3,19,2,6,2,270.062,270.062,0,1,0,0,0,0,1,1,0,Between -11 and 5
-161,Tazemetostat,572.7,83.1,992.0,4.2,42,2,6,9,572.336,572.336,0,1,0,0,0,0,0,0,0,Between -11 and 5
-162,Acalabrutinib,465.5,119.0,845.0,3.0,35,2,6,4,465.191,465.191,0,1,0,1,1,0,0,0,0,Between -11 and 5
-163,Ifenprodil,325.4,43.7,353.0,3.9,24,2,3,5,325.204,325.204,0,1,0,2,0,2,0,0,0,Between -11 and 5
-164,Ursodeoxycholic acid,392.6,77.8,605.0,4.9,28,3,4,4,392.293,392.293,0,1,0,10,10,0,0,0,0,Between -11 and 5
-165,Metoprolol succinate,652.8,176.0,308.0,,46,6,12,21,652.393,652.393,0,3,0,2,0,2,0,0,0,Larger than 6
-166,Merimepodib,452.5,124.0,652.0,2.1,33,3,7,8,452.17,452.17,0,1,0,1,1,0,0,0,0,Between -11 and 5
-167,Fludrocortisone acetate,422.5,101.0,838.0,1.7,30,2,7,4,422.21,422.21,0,1,0,7,7,0,0,0,0,Between -11 and 5
-168,Triazavirin,228.19,141.0,435.0,0.4,15,1,6,1,228.007,228.007,0,1,0,0,0,0,0,0,0,Between -11 and 5
-169,Ceftriaxone sodium,598.6,297.0,1120.0,,38,2,14,7,598.01,598.01,0,3,0,2,2,0,1,1,0,Larger than 6
-170,Rivaroxaban,435.9,116.0,645.0,2.5,29,1,6,5,435.066,435.066,0,1,0,1,1,0,0,0,0,Between -11 and 5
-171,Ventolin,337.39,156.0,309.0,,22,6,8,5,337.12,337.12,0,2,0,1,0,1,0,0,0,Larger than 6
-172,Apixaban,459.5,111.0,777.0,2.2,34,1,5,5,459.191,459.191,0,1,0,0,0,0,0,0,0,Between -11 and 5
-173,Ceftriaxone disodium salt hemiheptahydrate,1323.2,601.0,1120.0,,83,11,35,14,1322.09,1322.09,0,13,0,4,4,0,2,2,0,Larger than 6
-174,Vadadustat,306.7,99.5,393.0,2.5,21,3,5,4,306.041,306.041,0,1,0,0,0,0,0,0,0,Between -11 and 5
-175,Carbohydrate moiety of bromelain,1026.9,483.0,1680.0,-11.6,70,18,29,16,1026.38,1026.38,0,1,0,29,28,1,0,0,0,Smaller than -10
-176,Siponimod fumarate,1149.3,199.0,896.0,,82,4,20,20,1148.53,1148.53,0,3,0,0,0,0,3,3,0,Larger than 6
-177,(s)-1-(3-Chloro-4-fluorophenyl)ethanamine hydrochloride,210.07,26.0,131.0,,12,2,2,1,209.017,209.017,0,2,0,1,1,0,0,0,0,Larger than 6
-178,Prezcobix,1323.7,343.0,1980.0,,92,6,19,32,1322.59,1322.59,0,2,0,8,8,0,0,0,0,Larger than 6
-179,"[(2R,3R,4R,5R)-5-(4-amino-5-deuteriopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-bis(2-methylpropanoyloxy)oxolan-2-yl]methyl 2-methylpropanoate;hydrobromide",583.4,168.0,887.0,,37,2,11,11,582.155,582.155,0,2,1,4,4,0,0,0,0,Larger than 6
-180,Berberine,336.4,40.8,488.0,3.6,25,0,4,2,336.124,336.124,1,1,0,0,0,0,0,0,0,Between -11 and 5
-181,Cyproheptadine,287.4,3.2,423.0,4.7,22,0,1,0,287.167,287.167,0,1,0,0,0,0,0,0,0,Between -11 and 5
-182,Doxazosin,451.5,112.0,678.0,2.5,33,1,9,4,451.186,451.186,0,1,0,1,0,1,0,0,0,Between -11 and 5
-183,Fluconazole,306.27,81.6,358.0,0.4,22,1,7,5,306.104,306.104,0,1,0,0,0,0,0,0,0,Between -11 and 5
-184,Irbesartan,428.5,87.1,682.0,4.1,32,1,5,7,428.232,428.232,0,1,0,0,0,0,0,0,0,Between -11 and 5
-185,Sulfamethoxazole,253.28,107.0,346.0,0.9,17,2,6,3,253.052,253.052,0,1,0,0,0,0,0,0,0,Between -11 and 5
-186,D-Glucose,180.16,110.0,151.0,-2.6,12,5,6,1,180.063,180.063,0,1,0,5,4,1,0,0,0,Between -11 and 5
-187,N-Vinyl-2-pyrrolidone,111.14,20.3,120.0,0.4,8,0,1,1,111.068,111.068,0,1,0,0,0,0,0,0,0,Between -11 and 5
-188,Trimetazidine,266.34,43.0,259.0,1.0,19,1,5,5,266.163,266.163,0,1,0,0,0,0,0,0,0,Between -11 and 5
-189,Estetrol,304.4,80.9,441.0,1.5,22,4,4,0,304.167,304.167,0,1,0,7,7,0,0,0,0,Between -11 and 5
-190,Deferoxamine mesylate,656.8,269.0,832.0,,44,7,12,23,656.341,656.341,0,2,0,0,0,0,0,0,0,Larger than 6
-191,Tramadol Hydrochloride,299.83,32.7,282.0,,20,2,3,4,299.165,299.165,0,2,0,2,2,0,0,0,0,Larger than 6
-192,Nebivolol,405.4,71.0,483.0,3.0,29,3,7,6,405.175,405.175,0,1,0,4,0,4,0,0,0,Between -11 and 5
-193,Argatroban monohydrate,526.7,190.0,887.0,,36,6,9,9,526.257,526.257,0,2,0,4,3,1,0,0,0,Larger than 6
-194,Sulfamethoxazole and trimethoprim,543.6,212.0,653.0,,38,4,13,8,543.19,543.19,0,2,0,0,0,0,0,0,0,Larger than 6
-195,Zinc Gluconate,455.7,283.0,165.0,,27,10,14,8,454.03,454.03,0,3,0,8,8,0,0,0,0,Larger than 6
-196,"Zinc;(2R,3S,4R,5R)-2,3,4,5,6-pentahydroxyhexanoate",455.7,283.0,385.0,,27,10,14,8,454.03,454.03,0,3,0,8,8,0,0,0,0,Larger than 6
-197,(+)-Mefloquine,378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,2,0,0,0,0,Between -11 and 5
-198,"(2S,5R,6R)-6-[[(2S)-2-[(4-ethyl-2,3-dioxo-piperazine-1-carbonyl)amino]-2-phenyl-propanoyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid; (2S,3S,5R)-3-methyl-4,4,7-trioxo-3-(triazol-1-ylmethyl)-4$l^{6}-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid",831.9,313.0,1600.0,,57,4,15,9,831.232,831.232,0,2,0,7,7,0,0,0,0,Larger than 6
-199,Favipiravir,157.1,84.6,282.0,-0.6,11,2,4,1,157.029,157.029,0,1,0,0,0,0,0,0,0,Between -11 and 5
-200,Maraviroc,513.7,63.0,751.0,5.1,37,1,6,8,513.328,513.328,0,1,0,3,3,0,0,0,0,Larger than 6
-201,Toremifene,406.0,12.5,483.0,7.2,29,0,2,9,405.186,405.186,0,1,0,0,0,0,1,1,0,Larger than 6
-202,"(3-methyl-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)acetic acid",234.21,77.9,369.0,0.4,17,1,4,2,234.064,234.064,0,1,0,0,0,0,0,0,0,Between -11 and 5
-203,Enalapril maleate,492.5,171.0,638.0,,35,4,10,12,492.211,492.211,0,2,0,3,3,0,1,1,0,Larger than 6
-204,Naltrexone hydrochloride,377.9,70.0,621.0,,26,3,5,2,377.139,377.139,0,2,0,4,4,0,0,0,0,Larger than 6
-205,Chlorhexidine Gluconate,897.8,455.0,819.0,,60,18,16,23,896.32,896.32,0,3,0,8,8,0,2,2,0,Larger than 6
-206,Piperacillin/tazobactam,817.9,313.0,1550.0,,56,4,15,9,817.216,817.216,0,2,0,7,7,0,0,0,0,Larger than 6
-207,3-[5-(azetidine-1-carbonyl)pyrazin-2-yl]oxy-5-[(2S)-1-methoxypropan-2-yl]oxy-N-(5-methylpyrazin-2-yl)benzamide,478.5,129.0,710.0,1.3,35,1,9,9,478.196,478.196,0,1,0,1,1,0,0,0,0,Between -11 and 5
-208,Degarelix,1632.3,513.0,3390.0,3.5,117,17,18,41,1630.75,1630.75,0,1,0,11,11,0,0,0,0,Between -11 and 5
-209,(3S)-3-amino-6-(diaminomethylideneazaniumyl)hex-1-en-2-olate,172.23,115.0,174.0,-0.8,12,4,2,5,172.132,172.132,0,1,0,1,1,0,0,0,0,Between -11 and 5
-210,Sivelestat sodium,528.5,154.0,738.0,,35,6,12,9,528.139,528.139,0,6,0,0,0,0,0,0,0,Larger than 6
-211,CID 23679441,576.6,291.0,1120.0,,37,3,13,8,576.028,576.028,0,2,0,2,2,0,1,1,0,Larger than 6
-212,Masitinib mesylate,594.8,164.0,788.0,,41,3,10,7,594.208,594.208,0,2,0,0,0,0,0,0,0,Larger than 6
-213,"[(1S,4R,6S,7Z,18R)-4-(cyclopropylsulfonylcarbamoyl)-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-3,16-diazatricyclo[14.3.0.04,6]nonadec-7-en-18-yl] 4-fluoro-1,3-dihydroisoindole-2-carboxylate",731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,4,1,1,1,0,Between -11 and 5
-214,Natrii chloridi solutio composita,309.11,63.4,24.8,,13,4,8,0,307.852,307.852,0,10,0,0,0,0,0,0,0,Larger than 6
-215,CID 87060529,577.6,288.0,1110.0,,37,4,13,8,577.036,577.036,0,2,0,2,2,0,1,1,0,Larger than 6
-216,"(2S,3R)-3-[(2-aminopyridin-4-yl)methyl]-1-[[(1R)-1-cyclooctylethyl]carbamoyl]-4-oxoazetidine-2-carboxylic acid",402.5,126.0,606.0,3.7,29,3,6,5,402.227,402.227,0,1,0,3,3,0,0,0,0,Between -11 and 5
-217,Sinapultide acetate,2529.5,813.0,4910.0,,178,28,30,93,2528.85,2527.85,0,2,0,21,21,0,0,0,0,Larger than 6
-218,Bemnifosbuvir hemisulfate,1261.1,453.0,1000.0,,85,10,32,24,1260.4,1260.4,0,3,0,12,12,0,0,0,0,Larger than 6
-219,Reamberin,357.27,193.0,216.0,,23,6,10,7,357.101,357.101,0,4,0,4,4,0,0,0,0,Larger than 6
-220,Pomotrelvir,455.9,127.0,779.0,3.1,32,4,4,8,455.172,455.172,0,1,0,3,3,0,0,0,0,Between -11 and 5
-221,Selenious acid,128.99,57.5,26.3,,4,2,3,0,129.917,129.917,0,1,0,0,0,0,0,0,0,Larger than 6
-222,Spermidine,145.25,64.099,56.8,-1.0,10,3,3,7,145.158,145.158,0,1,0,0,0,0,0,0,0,Between -11 and 5
-223,Salbutamol,239.31,72.7,227.0,0.3,17,4,4,5,239.152,239.152,0,1,0,1,0,1,0,0,0,Between -11 and 5
-224,Bromhexine,376.13,29.3,256.0,4.3,18,1,2,3,375.997,373.999,0,1,0,0,0,0,0,0,0,Between -11 and 5
-225,Ebselen,274.19,20.3,275.0,,16,0,1,1,274.985,274.985,0,1,0,0,0,0,0,0,0,Larger than 6
-226,Fluoxetine,309.33,21.3,308.0,4.0,22,1,5,6,309.134,309.134,0,1,0,1,0,1,0,0,0,Between -11 and 5
-227,Ketotifen,309.4,48.6,476.0,3.2,22,0,3,0,309.119,309.119,0,1,0,0,0,0,0,0,0,Between -11 and 5
-228,Mefloquine,378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,0,2,0,0,0,Between -11 and 5
-229,Midazolam,325.8,30.2,471.0,2.5,23,0,3,1,325.078,325.078,0,1,0,0,0,0,0,0,0,Between -11 and 5
-230,Nitroglycerin,227.09,165.0,219.0,1.6,15,0,9,5,227.003,227.003,0,1,0,0,0,0,0,0,0,Between -11 and 5
-231,Quetiapine,383.5,73.6,496.0,2.1,27,1,5,6,383.167,383.167,0,1,0,0,0,0,0,0,0,Between -11 and 5
-232,Liothyronine,650.97,92.8,402.0,1.7,23,3,5,5,650.79,650.79,0,1,0,1,1,0,0,0,0,Between -11 and 5
-233,Pyridostigmine bromide,261.12,33.4,183.0,,14,0,3,2,260.016,260.016,0,2,0,0,0,0,0,0,0,Larger than 6
-234,Psilocybine,284.25,85.8,347.0,-1.6,19,3,5,5,284.093,284.093,0,1,0,0,0,0,0,0,0,Between -11 and 5
-235,Luminol,177.16,84.2,254.0,0.3,13,3,3,0,177.054,177.054,0,1,0,0,0,0,0,0,0,Between -11 and 5
-236,Acetylcysteine,163.2,67.4,148.0,0.4,10,3,4,3,163.03,163.03,0,1,0,1,1,0,0,0,0,Between -11 and 5
-237,Cyproheptadine hydrochloride,323.9,3.2,423.0,,23,1,1,0,323.144,323.144,0,2,0,0,0,0,0,0,0,Larger than 6
-238,Carmoisine,502.4,176.0,854.0,,33,1,9,2,501.988,501.988,0,3,0,0,0,0,0,0,0,Larger than 6
-239,Etoposide,588.6,161.0,969.0,0.6,42,3,13,5,588.184,588.184,0,1,0,10,10,0,0,0,0,Between -11 and 5
-240,Albuterol Sulfate,576.7,228.0,309.0,,39,10,12,10,576.272,576.272,0,3,0,2,0,2,0,0,0,Larger than 6
-241,Dexbudesonide,430.5,93.1,862.0,2.5,31,2,6,4,430.236,430.236,0,1,0,9,9,0,0,0,0,Between -11 and 5
-242,"2,2-Dimethyl-4-(chloromethyl)-1,3-dioxa-2-silacyclopentane",166.68,18.5,107.0,,9,0,2,1,166.022,166.022,0,1,0,1,0,1,0,0,0,Larger than 6
-243,Clopidogrel,321.8,57.8,381.0,3.8,21,0,4,4,321.059,321.059,0,1,0,1,1,0,0,0,0,Between -11 and 5
-244,Valsartan,435.5,112.0,608.0,4.4,32,2,6,10,435.227,435.227,0,1,0,1,1,0,0,0,0,Between -11 and 5
-245,Tirofiban,440.6,113.0,579.0,1.4,30,3,7,14,440.234,440.234,0,1,0,1,1,0,0,0,0,Between -11 and 5
-246,Voriconazole,349.31,76.7,448.0,1.5,25,1,8,5,349.115,349.115,0,1,0,2,2,0,0,0,0,Between -11 and 5
-247,N-Phenylethylenediamine,136.19,38.0,77.3,0.6,10,2,2,3,136.1,136.1,0,1,0,0,0,0,0,0,0,Between -11 and 5
-248,Oseltamivir phosphate,410.4,168.0,468.0,,27,5,9,8,410.182,410.182,0,2,0,3,3,0,0,0,0,Larger than 6
-249,Nicotine,162.23,16.1,147.0,1.2,12,0,2,1,162.116,162.116,0,1,0,1,1,0,0,0,0,Between -11 and 5
-250,Lactose monohydrate,360.31,191.0,382.0,,24,9,12,4,360.127,360.127,0,2,0,10,10,0,0,0,0,Larger than 6
-251,"2,4-Dioxaspiro[5.5]undec-8-ene, 3-(2-furanyl)-",220.26,31.6,266.0,2.1,16,0,3,1,220.11,220.11,0,1,0,0,0,0,0,0,0,Between -11 and 5
-252,Sivelestat,434.5,147.0,731.0,3.0,30,3,8,9,434.115,434.115,0,1,0,0,0,0,0,0,0,Between -11 and 5
-253,Tafenoquine,463.5,78.6,597.0,5.4,33,2,9,9,463.208,463.208,0,1,0,1,0,1,0,0,0,Larger than 6
-254,IB-Meca,510.3,134.0,589.0,0.9,29,4,8,5,510.051,510.051,0,1,0,4,4,0,0,0,0,Between -11 and 5
-255,"(S)-Hexahydropyrrolo[1,2-a]pyrazine-1,4-dione",154.17,49.4,215.0,-0.6,11,1,2,0,154.074,154.074,0,1,0,1,1,0,0,0,0,Between -11 and 5
-256,Arbidol,477.4,80.0,546.0,4.4,29,1,5,8,476.077,476.077,0,1,0,0,0,0,0,0,0,Between -11 and 5
-257,Tempol,172.24,24.5,159.0,0.9,12,1,2,0,172.134,172.134,0,1,0,0,0,0,0,0,0,Between -11 and 5
-258,Atazanavir,704.9,171.0,1110.0,5.6,51,5,9,18,704.39,704.39,0,1,0,4,4,0,0,0,0,Larger than 6
-259,Centhaquine,331.5,19.4,404.0,4.5,25,0,3,4,331.205,331.205,0,1,0,0,0,0,0,0,0,Between -11 and 5
-260,Regadenoson,390.35,187.0,587.0,-1.5,28,5,10,4,390.14,390.14,0,1,0,4,4,0,0,0,0,Between -11 and 5
-261,Teprotide,1101.3,387.0,2330.0,-0.9,79,10,13,24,1100.58,1100.58,0,1,0,10,10,0,0,0,0,Between -11 and 5
-262,Azithromycin,749.0,180.0,1150.0,4.0,52,5,14,7,748.509,748.509,0,1,0,18,18,0,0,0,0,Between -11 and 5
-263,Posaconazole,700.8,112.0,1170.0,4.6,51,1,11,12,700.33,700.33,0,1,0,4,4,0,0,0,0,Between -11 and 5
-264,Cannabidiol,314.5,40.5,414.0,6.5,23,2,2,6,314.225,314.225,0,1,0,2,2,0,0,0,0,Larger than 6
-265,"(R)-[2,8-bis(trifluoromethyl)-4-quinolyl]-[(2R)-2-piperidyl]methanol",378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,2,0,0,0,0,Between -11 and 5
-266,Tetramethylol-melamin-dioxy-propylen [German],358.35,170.0,377.0,-0.8,25,6,12,11,358.16,358.16,0,1,0,2,0,2,0,0,0,Between -11 and 5
-267,Heme arginate,792.7,260.0,1180.0,,55,8,10,13,792.305,792.305,2,3,0,1,1,0,0,0,0,Larger than 6
-268,Zincacetate,185.5,74.6,78.6,,9,2,4,0,183.971,183.971,0,3,0,0,0,0,0,0,0,Larger than 6
-269,Montelukast,586.2,95.7,891.0,7.7,41,2,5,12,585.21,585.21,0,1,0,1,1,0,1,1,0,Larger than 6
-270,Crocetin,328.4,74.6,608.0,5.4,24,2,4,8,328.167,328.167,0,1,0,0,0,0,7,7,0,Larger than 6
-271,Fondaparinux,1508.3,873.0,3450.0,-14.7,91,19,52,30,1506.95,1506.95,0,1,0,25,25,0,0,0,0,Smaller than -10
-272,Fluvoxamine,318.33,56.8,327.0,2.6,22,1,7,9,318.156,318.156,0,1,0,0,0,0,1,1,0,Between -11 and 5
-273,Naltrexone,341.4,70.0,621.0,1.9,25,2,5,2,341.163,341.163,0,1,0,4,4,0,0,0,0,Between -11 and 5
-274,20-Hydroxyecdysone,480.6,138.0,869.0,0.5,34,6,7,5,480.309,480.309,0,1,0,10,10,0,0,0,0,Between -11 and 5
-275,Ceftriaxone,554.6,288.0,1110.0,-1.3,36,4,13,8,554.046,554.046,0,1,0,2,2,0,1,1,0,Between -11 and 5
-276,"(3E,4S)-4-Hydroxy-3-{2-[(1R,4aS,5R,6R,8aS)-6-hydroxy-5-(hydroxymethyl)-5,8a-dimethyl-2-methylenedecahydronaphthalen-1-yl]ethylidene}dihydrofuran-2(3H)-one",350.4,87.0,597.0,2.2,25,3,5,3,350.209,350.209,0,1,0,6,2,4,1,1,0,Between -11 and 5
-277,Artemether and lumefantrine,827.3,69.6,1100.0,,56,1,7,11,825.333,825.333,0,2,0,9,7,2,1,1,0,Larger than 6
-278,Isavuconazonium,717.8,188.0,1210.0,4.1,51,2,13,15,717.242,717.242,1,1,0,3,2,1,0,0,0,Between -11 and 5
-279,Luminol sodium salt,200.15,84.2,254.0,,14,3,3,0,200.044,200.044,0,2,0,0,0,0,0,0,0,Larger than 6
-280,Dapagliflozin,408.9,99.4,472.0,2.3,28,4,6,6,408.134,408.134,0,1,0,5,5,0,0,0,0,Between -11 and 5
-281,Zinc Picolinate,309.6,106.0,108.0,,19,0,6,0,307.978,307.978,0,3,0,0,0,0,0,0,0,Larger than 6
-282,"Pregna-1,4-diene-3,20-dione,21-(3-carboxy-1-oxopropoxy)-11,17-dihydroxy-6-methyl-, monosodiumsalt, (6a,11b)-",497.5,138.0,981.0,,35,3,8,7,497.215,497.215,0,2,0,8,8,0,0,0,0,Larger than 6
-283,Vortioxetine,298.4,40.6,316.0,4.2,21,1,3,3,298.15,298.15,0,1,0,0,0,0,0,0,0,Between -11 and 5
-284,Enisamium iodide,354.19,33.0,241.0,,18,1,2,3,354.023,354.023,0,2,0,0,0,0,0,0,0,Larger than 6
-285,Cenicriviroc,696.9,105.0,1060.0,7.5,50,1,7,17,696.371,696.371,0,1,0,1,1,0,1,1,0,Larger than 6
-286,Apremilast,460.5,128.0,825.0,1.8,32,1,7,8,460.13,460.13,0,1,0,1,1,0,0,0,0,Between -11 and 5
-287,Empagliflozin,450.9,109.0,558.0,2.0,31,4,7,6,450.145,450.145,0,1,0,6,6,0,0,0,0,Between -11 and 5
-288,N6-ethanimidoyl-D-lysine,187.24,102.0,192.0,-3.1,13,3,4,6,187.132,187.132,0,1,0,1,1,0,0,0,0,Between -11 and 5
-289,Emricasan,569.5,151.0,934.0,3.6,40,4,11,11,569.179,569.179,0,1,0,2,2,0,0,0,0,Between -11 and 5
-290,Aviptadil Acetate,3344.9,1480.0,7510.0,-13.7,234,51,51,116,3343.74,3342.73,0,1,0,31,0,31,0,0,0,Smaller than -10
-291,Quinine sulfate dihydrate,782.9,176.0,538.0,,55,6,14,8,782.356,782.356,0,5,0,8,8,0,0,0,0,Larger than 6
-292,Hydrocortisone 21-hemisuccinate sodium salt,485.5,138.0,908.0,,34,3,8,7,485.215,485.215,0,2,0,7,7,0,0,0,0,Larger than 6
-293,Liothyronine sodium,672.95,95.6,408.0,,24,2,5,5,672.772,672.772,0,2,0,1,1,0,0,0,0,Larger than 6
-294,sodium;8-amino-4-oxo-3H-phthalazin-1-olate,199.14,90.5,269.0,,14,2,4,0,199.036,199.036,0,2,0,0,0,0,0,0,0,Larger than 6
-295,Edoxaban tosylate monohydrate,738.3,229.0,1090.0,,49,5,12,6,737.207,737.207,0,3,0,3,3,0,0,0,0,Larger than 6
-296,Daclatasvir,738.9,175.0,1190.0,5.1,54,4,8,13,738.385,738.385,0,1,0,4,4,0,0,0,0,Larger than 6
-297,"2-[(4S)-4-amino-5,5-dihydroxyhexyl]guanidine",190.24,131.0,177.0,-2.5,13,5,4,5,190.143,190.143,0,1,0,1,1,0,0,0,0,Between -11 and 5
-298,Fosmanogepix,468.4,148.0,644.0,1.6,33,2,9,9,468.12,468.12,0,1,0,0,0,0,0,0,0,Between -11 and 5
-299,Larazotide acetate,785.9,339.0,1320.0,,55,10,13,21,785.428,785.428,0,2,0,5,5,0,0,0,0,Larger than 6
-300,Thymosin,3051.3,1370.0,6930.0,-22.0,212,47,57,109,3050.5,3049.5,0,1,0,30,30,0,0,0,0,Smaller than -10
-301,Sofosbuvir,529.5,153.0,913.0,1.0,36,3,11,11,529.163,529.163,0,1,0,6,6,0,0,0,0,Between -11 and 5
-302,Razuprotafib,586.7,212.0,906.0,3.7,39,4,10,12,586.101,586.101,0,1,0,2,2,0,0,0,0,Between -11 and 5
-303,Nalpha-[(4-Methylpiperazin-1-Yl)carbonyl]-N-[(3s)-1-Phenyl-5-(Phenylsulfonyl)pentan-3-Yl]-L-Phenylalaninamide,576.8,107.0,897.0,4.4,41,2,5,12,576.277,576.277,0,1,0,2,2,0,0,0,0,Between -11 and 5
-304,Ascorbic Acid,176.12,107.0,232.0,-1.6,12,4,6,2,176.032,176.032,0,1,0,2,2,0,0,0,0,Between -11 and 5
-305,Doxycycline,444.4,182.0,956.0,-0.7,32,6,9,2,444.153,444.153,0,1,0,6,6,0,0,0,0,Between -11 and 5
-306,Rabeximod,409.9,63.0,590.0,3.9,29,1,4,5,409.167,409.167,0,1,0,0,0,0,0,0,0,Between -11 and 5
-307,Kaolin,258.16,98.0,167.0,,13,2,9,4,257.902,257.902,0,3,0,0,0,0,0,0,0,Larger than 6
-308,H-Ile-OH.H-Thr-OH.H-Leu-OH.H-Val-OH.H-Met-OH.H-Phe-OH.H-Trp-OH.H-Lys-OH,1163.4,594.0,988.0,,80,19,27,25,1162.65,1162.65,0,8,0,10,10,0,0,0,0,Larger than 6
-309,Semaglutide,4114.0,1650.0,9590.0,-5.8,291,57,63,151,4112.12,4111.12,0,1,0,30,30,0,0,0,0,Between -11 and 5
-310,Isuzinaxib hydrochloride,315.8,45.2,412.0,,22,2,3,4,315.114,315.114,0,2,0,0,0,0,0,0,0,Larger than 6
-311,CID 66726979,457.5,147.0,731.0,,31,3,8,9,457.105,457.105,0,2,0,0,0,0,0,0,0,Larger than 6
-312,Desidustat,332.31,116.0,583.0,1.9,24,3,6,6,332.101,332.101,0,1,0,0,0,0,0,0,0,Between -11 and 5
-313,"7-[[2-Ethoxyimino-2-[5-(phosphonoamino)-1,2,4-thiadiazol-3-yl]acetyl]amino]-3-[[4-(1-methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid;acetate",744.7,368.0,1240.0,,47,5,19,11,744.031,744.031,0,2,0,2,0,2,1,0,1,Larger than 6
-314,1-(4-(((6-Amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)-4-fluoropiperidin-1-yl)prop-2-en-1-one,447.5,93.4,643.0,4.1,33,2,7,7,447.207,447.207,0,1,0,0,0,0,0,0,0,Between -11 and 5
-315,Ceftriaxone disodium hemiheptahydrate,600.6,288.0,1110.0,,38,4,13,8,600.026,600.026,0,3,0,2,2,0,1,1,0,Larger than 6
-316,Descovy,723.7,257.0,1050.0,,49,4,15,14,723.236,723.236,0,2,0,5,5,0,0,0,0,Larger than 6
-317,Zimlovisertib,361.4,104.0,535.0,2.0,26,2,6,6,361.144,361.144,0,1,0,3,3,0,0,0,0,Between -11 and 5
-318,CID 131673872,202.17,84.2,254.0,,14,3,3,0,202.059,202.059,0,3,0,0,0,0,0,0,0,Larger than 6
-319,"Sodium;5-amino-2,3-dihydrophthalazine-1,4-dione;hydride",201.16,84.2,254.0,,14,3,4,0,201.051,201.051,0,3,0,0,0,0,0,0,0,Larger than 6
-320,Zilucoplan,3562.0,1070.0,6980.0,4.8,251,28,57,142,3560.97,3559.97,0,1,0,16,16,0,0,0,0,Between -11 and 5
-321,Defibrotide,444.4,137.0,773.0,1.8,31,4,7,5,444.12,444.12,0,1,0,0,0,0,0,0,0,Between -11 and 5
-322,"7-[(3s,4r)-4-(3-Chlorophenyl)carbonylpyrrolidin-3-Yl]-3h-Quinazolin-4-One",353.8,70.6,567.0,2.1,25,2,4,3,353.093,353.093,0,1,0,2,2,0,0,0,0,Between -11 and 5
-323,Unii-7kyp9tkt70,879.6,484.0,1210.0,,58,15,24,13,879.205,879.205,0,4,0,8,0,8,0,0,0,Larger than 6
-324,"4-acetamidobenzoic acid;9-[(2R,3R,4R,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1H-purin-6-one;1-(dimethylamino)propan-2-ol",1115.2,399.0,658.0,,79,13,22,14,1114.55,1114.55,0,7,0,7,4,3,0,0,0,Larger than 6
-325,"[(1S,4R,6S)-4-(cyclopropylsulfonylcarbamoyl)-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-3,16-diazatricyclo[14.3.0.04,6]nonadec-7-en-18-yl] 4-fluoro-1,3-dihydroisoindole-2-carboxylate",731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,3,2,1,0,1,Between -11 and 5
-326,example 13 [US20210284598A1],257.279,40.5,287.0,2.7,18,1,4,4,257.123,257.123,0,1,0,0,0,0,0,0,0,Between -11 and 5
-327,Stannous protoporphyrin,679.4,102.0,1010.0,,43,2,8,8,680.145,680.145,0,2,0,0,0,0,0,0,0,Larger than 6
-328,"3,6-Di-O-acetyl-2-deoxy-d-glucopyranose",248.23,110.0,276.0,-1.7,17,2,7,9,248.09,248.09,0,1,0,3,3,0,0,0,0,Between -11 and 5
-329,X6 hydrobromide [PMID: 34584244],502.5,168.0,887.0,2.3,36,1,11,11,502.229,502.229,0,1,1,4,4,0,0,0,0,Between -11 and 5
-330,Enoxaparin,1134.9,652.0,2410.0,-10.8,70,15,38,21,1134.01,1134.01,0,1,0,20,0,20,0,0,0,Smaller than -10
-331,Amantadine,151.25,26.0,144.0,2.4,11,1,1,0,151.136,151.136,0,1,0,0,0,0,0,0,0,Between -11 and 5
-332,Amlodipine,408.9,99.9,647.0,3.0,28,2,7,10,408.145,408.145,0,1,0,1,0,1,0,0,0,Between -11 and 5
-333,Bicalutamide,430.4,116.0,750.0,2.3,29,2,9,5,430.061,430.061,0,1,0,1,0,1,0,0,0,Between -11 and 5
-334,Candesartan cilexetil,610.7,143.0,962.0,7.0,45,1,10,13,610.254,610.254,0,1,0,1,0,1,0,0,0,Larger than 6
-335,Formoterol,344.4,90.8,388.0,1.8,25,4,5,8,344.174,344.174,0,1,0,2,0,2,0,0,0,Between -11 and 5
-336,Hydroxychloroquine,335.9,48.4,331.0,3.6,23,2,4,9,335.176,335.176,0,1,0,1,0,1,0,0,0,Between -11 and 5
-337,Ibudilast,230.31,34.4,288.0,3.0,17,0,2,3,230.142,230.142,0,1,0,0,0,0,0,0,0,Between -11 and 5
-338,Lidocaine,234.34,32.299,228.0,2.3,17,1,2,5,234.173,234.173,0,1,0,0,0,0,0,0,0,Between -11 and 5
-339,Modafinil,273.4,79.4,302.0,1.7,19,1,3,5,273.082,273.082,0,1,0,1,0,1,0,0,0,Between -11 and 5
-340,Omeprazole,345.4,96.3,453.0,2.2,24,1,6,5,345.115,345.115,0,1,0,1,0,1,0,0,0,Between -11 and 5
-341,Pentoxifylline,278.31,75.5,426.0,0.3,20,0,4,5,278.138,278.138,0,1,0,0,0,0,0,0,0,Between -11 and 5
-342,Arginine,174.2,128.0,176.0,-4.2,12,4,4,5,174.112,174.112,0,1,0,1,1,0,0,0,0,Between -11 and 5
-343,"4,4'-Diphenylmethane diisocyanate",250.25,58.9,332.0,5.4,19,0,4,4,250.074,250.074,0,1,0,0,0,0,0,0,0,Larger than 6
-344,Carvacrol,150.22,20.2,120.0,3.1,11,1,1,1,150.104,150.104,0,1,0,0,0,0,0,0,0,Between -11 and 5
-345,Silver,107.868,0.0,0.0,,1,0,0,0,106.905,106.905,0,1,0,0,0,0,0,0,0,Larger than 6
-346,Phorbol 12-myristate 13-acetate,616.8,130.0,1150.0,6.5,44,3,8,17,616.398,616.398,0,1,0,8,8,0,0,0,0,Larger than 6
-347,(-)-Mefloquine,378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,2,0,0,0,0,Between -11 and 5
-348,Atorvastatin,558.6,112.0,822.0,5.0,41,4,6,12,558.253,558.253,0,1,0,2,2,0,0,0,0,Between -11 and 5
-349,Propranolol Hydrochloride,295.8,41.5,257.0,,20,3,3,6,295.134,295.134,0,2,0,1,0,1,0,0,0,Larger than 6
-350,Oseltamivir,312.4,90.6,418.0,1.1,22,2,5,8,312.205,312.205,0,1,0,3,3,0,0,0,0,Between -11 and 5
-351,"2,4,6-Trinitro-m-xylene",241.16,138.0,317.0,2.0,17,0,6,0,241.033,241.033,0,1,0,0,0,0,0,0,0,Between -11 and 5
-352,Argatroban,508.6,189.0,887.0,1.3,35,5,8,9,508.247,508.247,0,1,0,4,3,1,0,0,0,Between -11 and 5
-353,Lopinavir,628.8,120.0,940.0,5.9,46,4,5,15,628.362,628.362,0,1,0,4,4,0,0,0,0,Larger than 6
-354,Allopregnanolone,318.5,37.3,500.0,4.9,23,1,2,1,318.256,318.256,0,1,0,8,8,0,0,0,0,Between -11 and 5
-355,Fingolimod,307.5,66.5,258.0,4.2,22,3,3,12,307.251,307.251,0,1,0,0,0,0,0,0,0,Between -11 and 5
-356,Imatinib Mesylate,589.7,149.0,799.0,,42,3,10,7,589.247,589.247,0,2,0,0,0,0,0,0,0,Larger than 6
-357,Meldonium,146.19,52.2,112.0,-2.1,10,1,3,3,146.106,146.106,0,1,0,0,0,0,0,0,0,Between -11 and 5
-358,Ramatroban,416.5,96.8,689.0,2.9,29,2,6,6,416.121,416.121,0,1,0,1,1,0,0,0,0,Between -11 and 5
-359,Ivabradine,468.6,60.5,663.0,2.4,34,0,6,10,468.262,468.262,0,1,0,1,1,0,0,0,0,Between -11 and 5
-360,Moxifloxacin,401.4,82.1,727.0,0.6,29,2,8,4,401.175,401.175,0,1,0,2,2,0,0,0,0,Between -11 and 5
-361,Varespladib,380.4,112.0,589.0,2.8,28,2,5,8,380.137,380.137,0,1,0,0,0,0,0,0,0,Between -11 and 5
-362,Naproxen,230.26,46.5,277.0,3.3,17,1,3,3,230.094,230.094,0,1,0,1,1,0,0,0,0,Between -11 and 5
-363,Dabigatran,471.5,150.0,757.0,1.7,35,4,7,9,471.202,471.202,0,1,0,0,0,0,0,0,0,Between -11 and 5
-364,Senicapoc,323.3,43.1,397.0,4.1,24,1,3,4,323.112,323.112,0,1,0,0,0,0,0,0,0,Between -11 and 5
-365,Povidone iodine,364.95,20.3,120.0,,10,0,1,1,364.877,364.877,0,2,0,0,0,0,0,0,0,Larger than 6
-366,beta-L-Arabinose,150.13,90.2,117.0,-2.5,10,4,5,0,150.053,150.053,0,1,0,4,4,0,0,0,0,Between -11 and 5
-367,Quinidine,324.4,45.6,457.0,2.9,24,1,4,4,324.184,324.184,0,1,0,4,4,0,0,0,0,Between -11 and 5
-368,Pectin,194.14,127.0,205.0,-2.3,13,5,7,1,194.043,194.043,0,1,0,5,5,0,0,0,0,Between -11 and 5
-369,Fluticasone Propionate,500.6,106.0,984.0,4.0,34,1,9,6,500.184,500.184,0,1,0,9,9,0,0,0,0,Between -11 and 5
-370,Decitabine,228.21,121.0,356.0,-1.2,16,3,4,2,228.086,228.086,0,1,0,3,3,0,0,0,0,Between -11 and 5
-371,Bucillamine,223.3,68.4,218.0,0.4,13,4,5,4,223.034,223.034,0,1,0,1,1,0,0,0,0,Between -11 and 5
-372,Canrenoic acid,358.5,74.6,707.0,1.9,26,2,4,3,358.214,358.214,0,1,0,6,6,0,0,0,0,Between -11 and 5
-373,"(S)-[2,8-bis(trifluoromethyl)quinolin-4-yl]-[(2S)-piperidin-2-yl]methanol",378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,2,0,0,0,0,Between -11 and 5
-374,"(-)-(S)-9-Fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyrido(1,2,3-de)-1,4-benzoxazine-6-carboxylic acid, hemihydrate",740.7,148.0,634.0,,53,3,17,4,740.298,740.298,0,3,0,2,2,0,0,0,0,Larger than 6
-375,"(S,S)-Formoterol",344.4,90.8,388.0,1.8,25,4,5,8,344.174,344.174,0,1,0,2,2,0,0,0,0,Between -11 and 5
-376,Glatiramer acetate,623.7,374.0,519.0,,43,12,18,13,623.301,623.301,0,5,0,4,4,0,0,0,0,Larger than 6
-377,Isoquercetin,464.4,207.0,758.0,0.4,33,8,12,4,464.095,464.095,0,1,0,5,5,0,0,0,0,Between -11 and 5
-378,Pitavastatin,421.5,90.6,631.0,3.5,31,3,6,8,421.169,421.169,0,1,0,2,2,0,1,1,0,Between -11 and 5
-379,Deoxy-methyl-arginine,172.23,108.0,174.0,-1.8,12,3,3,5,172.132,172.132,0,1,0,1,1,0,0,0,0,Between -11 and 5
-380,Dexmedetomidine,200.28,28.7,205.0,3.1,15,1,1,2,200.131,200.131,0,1,0,1,1,0,0,0,0,Between -11 and 5
-381,Nafamostat mesylate,539.6,266.0,645.0,,36,6,10,5,539.114,539.114,0,3,0,0,0,0,0,0,0,Larger than 6
-382,Bromhexine Hydrochloride,412.59,29.3,256.0,,19,2,2,3,411.974,409.976,0,2,0,0,0,0,0,0,0,Larger than 6
-383,Tenofovir Disoproxil Fumarate,635.5,260.0,817.0,,43,3,18,19,635.184,635.184,0,2,0,1,1,0,1,1,0,Larger than 6
-384,Eritoran,1313.7,294.0,1900.0,15.4,89,7,19,59,1312.84,1312.84,0,1,0,11,11,0,1,1,0,Larger than 6
-385,Icatibant acetate,1364.6,589.0,2750.0,,96,16,20,30,1363.68,1363.68,0,2,0,12,12,0,0,0,0,Larger than 6
-386,Dalcetrapib,389.6,71.5,481.0,7.1,27,1,3,9,389.239,389.239,0,1,0,0,0,0,0,0,0,Larger than 6
-387,"7-[[(2Z)-2-ethoxyimino-2-[5-(phosphonoamino)-1,2,4-thiadiazol-3-yl]acetyl]amino]-3-[[4-(1-methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid",685.7,328.0,1220.0,1.6,43,5,17,11,685.018,685.018,1,1,0,2,0,2,1,1,0,Between -11 and 5
-388,Icosapent ethyl,330.5,26.3,425.0,6.3,24,0,2,15,330.256,330.256,0,1,0,0,0,0,5,5,0,Larger than 6
-389,"1-Piperazinecarboxamide, 4-methyl-N-((1S)-2-oxo-2-(((1S)-1-(2-phenylethyl)-3-(phenylsulfonyl)-2-propenyl)amino)-1-(phenylmethyl)ethyl)-",574.7,107.0,939.0,4.1,41,2,5,11,574.261,574.261,0,1,0,2,2,0,1,1,0,Between -11 and 5
-390,Remimazolam,439.3,69.4,601.0,3.4,28,0,5,5,438.069,438.069,0,1,0,1,1,0,0,0,0,Between -11 and 5
-391,Azithromycin Monohydrate,767.0,181.0,1150.0,,53,6,15,7,766.519,766.519,0,2,0,18,18,0,0,0,0,Larger than 6
-392,Danoprevir,731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,5,0,1,1,0,Between -11 and 5
-393,Mitoquinone,583.7,52.6,886.0,9.4,42,0,4,16,583.298,583.298,1,1,0,0,0,0,0,0,0,Larger than 6
-394,Belnacasan,509.0,140.0,818.0,2.3,35,3,7,8,508.209,508.209,0,1,0,4,4,0,0,0,0,Between -11 and 5
-395,Apilimod mesylate,610.7,210.0,637.0,,41,3,14,8,610.188,610.188,0,3,0,0,0,0,1,1,0,Larger than 6
-396,Losmapimod,383.5,71.1,573.0,3.8,28,2,4,6,383.201,383.201,0,1,0,0,0,0,0,0,0,Between -11 and 5
-397,Emtricitabine and tenofovir disoproxil fumarate,882.8,374.0,1190.0,,59,5,23,21,882.227,882.227,0,3,0,3,3,0,1,1,0,Larger than 6
-398,Dapansutrile,133.17,66.3,190.0,-0.7,8,0,3,2,133.02,133.02,0,1,0,0,0,0,0,0,0,Between -11 and 5
-399,"(S)-[2,8-bis(trifluoromethyl)quinolin-4-yl]-piperidin-2-ylmethanol",378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,1,1,0,0,0,Between -11 and 5
-400,Tridecactide,1623.8,659.0,3240.0,-6.2,115,23,24,50,1622.77,1622.77,0,1,0,12,12,0,0,0,0,Between -11 and 5
-401,Aviptadil,3326.8,1470.0,7580.0,-15.9,234,51,51,115,3325.74,3324.74,0,1,0,31,31,0,0,0,0,Smaller than -10
-402,Pamapimod,406.4,108.0,591.0,2.4,29,3,9,8,406.145,406.145,0,1,0,0,0,0,0,0,0,Between -11 and 5
-403,Sodium pyruvate,110.04,57.2,88.2,,7,0,3,1,109.998,109.998,0,2,0,0,0,0,0,0,0,Larger than 6
-404,Brequinar sodium,397.3,53.0,557.0,,29,0,5,3,397.089,397.089,0,2,0,0,0,0,0,0,0,Larger than 6
-405,Montelukast Sodium,608.2,98.6,898.0,,42,1,5,12,607.192,607.192,0,2,0,1,1,0,1,1,0,Larger than 6
-406,Sivelestat sodium anhydrous,456.4,150.0,738.0,,31,2,8,9,456.097,456.097,0,2,0,0,0,0,0,0,0,Larger than 6
-407,Methylprednisolone sodium succinate,496.5,141.0,988.0,,35,2,8,7,496.207,496.207,0,2,0,8,8,0,0,0,0,Larger than 6
-408,Bromelains,248.25,81.6,344.0,,17,1,3,4,248.114,248.114,0,2,0,2,0,2,0,0,0,Larger than 6
-409,Fostamatinib disodium,732.5,198.0,893.0,,48,8,21,9,732.176,732.176,0,9,0,0,0,0,0,0,0,Larger than 6
-410,Phosphate-Buffered Saline,411.04,164.0,96.3,,17,3,10,0,409.765,409.765,0,9,0,0,0,0,0,0,0,Larger than 6
-411,4'-C-Azido-2'-deoxy-2'-fluoro-b-D-arabinocytidine,286.22,123.0,533.0,-0.8,20,3,7,3,286.083,286.083,0,1,0,4,0,4,1,0,1,Between -11 and 5
-412,5-Chloro-2-([(2-([3-(furan-2-yl)phenyl]amino)-2-oxoethoxy)acetyl]amino)benzoic acid,428.8,118.0,618.0,3.3,30,3,6,8,428.078,428.078,0,1,0,0,0,0,0,0,0,Between -11 and 5
-413,"[(2R,3S,4R)-4-acetyloxy-3,6-dihydroxyoxan-2-yl]methyl acetate",248.23,102.0,290.0,-1.0,17,2,7,5,248.09,248.09,0,1,0,4,3,1,0,0,0,Between -11 and 5
-414,Ketone Ester,176.21,66.8,135.0,-0.1,12,2,4,6,176.105,176.105,0,1,0,2,2,0,0,0,0,Between -11 and 5
-415,CID 45114162,520.6,152.0,761.0,,34,2,10,9,520.123,520.123,1,2,0,2,2,0,1,1,0,Larger than 6
-416,Alvelestat,545.5,123.0,1100.0,2.5,38,1,9,6,545.134,545.134,0,1,0,0,0,0,0,0,0,Between -11 and 5
-417,Emvododstat,467.3,54.6,651.0,6.3,32,1,3,4,466.085,466.085,0,1,0,1,1,0,0,0,0,Larger than 6
-418,8-chloro-N-[4-(trifluoromethoxy)phenyl]quinolin-2-amine,338.71,34.2,388.0,5.9,23,1,6,3,338.043,338.043,0,1,0,0,0,0,0,0,0,Larger than 6
-419,Riamilovir sodium dihydrate,286.2,152.0,262.0,,18,2,10,1,286.01,286.01,0,4,0,0,0,0,0,0,0,Larger than 6
-420,Doxycycline hyclate,545.0,203.0,958.0,,37,9,11,2,544.182,544.182,0,4,0,6,6,0,0,0,0,Larger than 6
-421,Budesonide and formoterol fumarate dihydrate,774.9,184.0,1250.0,,56,6,11,12,774.409,774.409,0,2,0,11,10,1,0,0,0,Larger than 6
-422,Vafidemstat,336.4,86.2,410.0,2.2,25,2,6,7,336.159,336.159,0,1,0,2,2,0,0,0,0,Between -11 and 5
-423,Ledipasvir,889.0,175.0,1820.0,7.4,65,4,10,12,888.413,888.413,0,1,0,6,6,0,0,0,0,Larger than 6
-424,Telacebec,557.0,58.9,796.0,7.9,39,1,7,7,556.185,556.185,0,1,0,0,0,0,0,0,0,Larger than 6
-425,Acebilustat,481.5,79.0,728.0,2.2,36,1,7,8,481.2,481.2,0,1,0,2,2,0,0,0,0,Between -11 and 5
-426,Quinidine monohydrate,342.4,46.6,457.0,,25,2,5,4,342.194,342.194,0,2,0,4,4,0,0,0,0,Larger than 6
-427,Dexamethasone phosphate disodium,518.4,141.0,973.0,,34,4,9,4,518.146,518.146,0,3,0,8,8,0,0,0,0,Larger than 6
-428,Montmorillonite,360.31,141.0,18.3,,18,1,12,0,359.825,359.825,0,10,0,0,0,0,0,0,0,Larger than 6
-429,methyl N-[(2R)-2-[2-[5-[4-[4-[2-[1-[(2S)-2-(methoxycarbonylamino)-3-methyl-butanoyl]pyrrolidin-2-yl]-1H-imidazol-5-yl]phenyl]phenyl]-1H-imidazol-2-yl]pyrrolidine-1-carbonyl]-3-methyl-butyl]carbamate,752.9,175.0,1300.0,5.0,55,4,8,14,752.401,752.401,0,1,0,4,2,2,0,0,0,Between -11 and 5
-430,Nadide sodium,685.4,324.0,1110.0,,45,6,18,11,685.091,685.091,0,2,0,8,8,0,0,0,0,Larger than 6
-431,"(6R,7R)-7-[[(2E)-2-ethoxyimino-2-[5-(phosphonoamino)-1,2,4-thiadiazol-3-yl]acetyl]amino]-3-[[4-(1-methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate",684.7,330.0,1210.0,2.3,43,4,17,10,684.01,684.01,0,1,0,2,2,0,1,1,0,Between -11 and 5
-432,"3-Deoxy-3-[4-(3-Fluorophenyl)-1h-1,2,3-Triazol-1-Yl]-Beta-D-Galactopyranosyl 3-Deoxy-3-[4-(3-Fluorophenyl)-1h-1,2,3-Triazol-1-Yl]-1-Thio-Beta-D-Galactopyranoside",648.6,227.0,903.0,0.2,45,6,15,8,648.181,648.181,0,1,0,10,10,0,0,0,0,Between -11 and 5
-433,"N-[(2S,3S,4R)-1-(alpha-D-galactopyranosyloxy)-3,4-dihydroxyoctadecan-2-yl]undecanamide",647.9,169.0,700.0,7.9,45,7,9,29,647.497,647.497,0,1,0,8,8,0,0,0,0,Larger than 6
-434,Brensocatib,420.5,104.0,699.0,2.0,31,2,6,5,420.18,420.18,0,1,0,2,2,0,0,0,0,Between -11 and 5
-435,Ezurpimtrostat,423.0,40.2,520.0,5.9,30,2,4,6,422.224,422.224,0,1,0,0,0,0,0,0,0,Larger than 6
-436,Dexamethasone 21-phosphate disodium salt,548.5,141.0,973.0,,36,4,9,4,548.193,548.193,0,4,0,8,8,0,0,0,0,Larger than 6
-437,Enpatoran,320.31,65.9,472.0,2.7,23,1,7,1,320.125,320.125,0,1,0,2,2,0,0,0,0,Between -11 and 5
-438,H-D-Lys-D-Leu-D-Leu-D-Leu-D-Leu-D-Lys-D-Leu-D-Leu-D-Leu-D-Leu-D-Lys-D-Leu-D-Leu-D-Leu-D-Leu-D-Lys-D-Leu-D-Leu-D-Leu-D-Leu-D-Lys-OH,2469.4,775.0,4880.0,11.9,174,27,28,93,2468.83,2467.83,0,1,0,21,21,0,0,0,0,Larger than 6
-439,CID 131844884,1738.2,873.0,3450.0,,101,19,52,30,1736.85,1736.85,0,11,0,25,25,0,0,0,0,Larger than 6
-440,Folic Acid,441.4,209.0,767.0,-1.1,32,6,10,9,441.14,441.14,0,1,0,1,1,0,0,0,0,Between -11 and 5
-441,Inosine pranobex,1115.2,399.0,658.0,,79,13,22,14,1114.55,1114.55,0,7,0,7,4,3,0,0,0,Larger than 6
-442,Eltrombopag,442.5,115.0,812.0,5.4,33,3,7,5,442.164,442.164,0,1,0,0,0,0,0,0,0,Larger than 6
-443,Rintatolimod,995.6,517.0,1600.0,,65,15,27,12,995.135,995.135,0,3,0,12,12,0,0,0,0,Larger than 6
-444,Normosang,792.7,260.0,1180.0,,55,8,10,13,792.305,792.305,2,3,0,1,1,0,0,0,0,Larger than 6
-445,Artecom,1294.4,485.0,1380.0,,84,15,31,12,1293.31,1293.31,0,6,0,8,8,0,0,0,0,Larger than 6
-446,Imunovir; Delimmun; Groprinosin;Inosine pranobex,1115.2,399.0,658.0,,79,13,22,14,1114.55,1114.55,0,7,0,7,0,7,0,0,0,Larger than 6
-447,"1-(4-fluorobenzene-6-id-1-yl)-N-phenylmethanimine;iridium(3+);1,2,3,4,5-pentamethylcyclopenta-1,3-diene;chloride",561.1,12.4,601.0,,27,0,5,1,561.121,561.121,0,4,0,0,0,0,0,0,0,Larger than 6
-448,EIDD-2801,329.31,141.0,534.0,-0.8,23,4,7,6,329.122,329.122,0,1,0,4,4,0,0,0,0,Between -11 and 5
-449,CID 154701548,287.21,143.0,435.0,,18,3,8,1,287.017,287.017,0,4,0,0,0,0,0,0,0,Larger than 6
-450,"alpha-D-Glucopyranoside, methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-alpha-D-glucopyranosyl-(1-->4)-O-beta-D-glucopyranuronosyl-(1-->4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-alpha-D-glucopyranosyl-(1-->4)-O-2-O-sulfo-alpha-L-idopyranuronosyl-(1-->4)-2-deoxy-2-(sulfoamino)-, 6-(hydrogen sulfate), sodium salt (1:10)",1531.3,873.0,3450.0,,92,19,52,30,1529.94,1529.94,0,2,0,25,25,0,0,0,0,Larger than 6
-451,"propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-[2-amino-6-(methylamino)purin-9-yl]-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate;sulfuric acid",679.6,268.0,1000.0,,45,6,18,12,679.184,679.184,0,2,0,6,5,1,0,0,0,Larger than 6
-452,Ensitrelvir,531.9,114.0,919.0,2.5,37,1,8,6,531.115,531.115,0,1,0,0,0,0,0,0,0,Between -11 and 5
-453,"3,4-Methylenedioxymethamphetamine",193.24,30.5,186.0,2.2,14,1,3,3,193.11,193.11,0,1,0,1,0,1,0,0,0,Between -11 and 5
-454,Acetaminophen,151.16,49.3,139.0,0.5,11,2,2,1,151.063,151.063,0,1,0,0,0,0,0,0,0,Between -11 and 5
-455,Amiodarone,645.3,42.7,547.0,7.6,31,0,4,11,645.024,645.024,0,1,0,0,0,0,0,0,0,Larger than 6
-456,Verapamil,454.6,64.0,606.0,3.8,33,0,6,13,454.283,454.283,0,1,0,1,0,1,0,0,0,Between -11 and 5
-457,Candesartan,440.5,119.0,660.0,4.1,33,2,7,7,440.16,440.16,0,1,0,0,0,0,0,0,0,Between -11 and 5
-458,Chlordiazepoxide,299.75,48.2,580.0,2.4,21,1,3,1,299.083,299.083,0,1,0,0,0,0,0,0,0,Between -11 and 5
-459,Chloroquine,319.9,28.2,309.0,4.6,22,1,3,8,319.182,319.182,0,1,0,1,0,1,0,0,0,Between -11 and 5
-460,Deferoxamine,560.7,206.0,739.0,-2.1,39,6,9,23,560.353,560.353,0,1,0,0,0,0,0,0,0,Between -11 and 5
-461,Famotidine,337.5,238.0,469.0,-0.6,20,4,8,7,337.045,337.045,0,1,0,0,0,0,1,0,1,Between -11 and 5
-462,Fenofibrate,360.8,52.6,458.0,5.2,25,0,4,7,360.113,360.113,0,1,0,0,0,0,0,0,0,Larger than 6
-463,Ketamine,237.72,29.1,269.0,2.2,16,1,2,2,237.092,237.092,0,1,0,1,0,1,0,0,0,Between -11 and 5
-464,Lansoprazole,369.4,87.1,480.0,2.8,25,1,8,5,369.076,369.076,0,1,0,1,0,1,0,0,0,Between -11 and 5
-465,Metformin,129.16,91.5,132.0,-1.3,9,3,1,2,129.101,129.101,0,1,0,0,0,0,0,0,0,Between -11 and 5
-466,Nafamostat,347.4,141.0,552.0,2.0,26,4,4,5,347.138,347.138,0,1,0,0,0,0,0,0,0,Between -11 and 5
-467,Palmitoylethanolamide,299.5,49.3,219.0,6.2,21,2,2,16,299.282,299.282,0,1,0,0,0,0,0,0,0,Larger than 6
-468,Pioglitazone,356.4,93.6,466.0,3.8,25,1,5,7,356.119,356.119,0,1,0,1,0,1,0,0,0,Between -11 and 5
-469,Propofol,178.27,20.2,135.0,3.8,13,1,1,2,178.136,178.136,0,1,0,0,0,0,0,0,0,Between -11 and 5
-470,Sevoflurane,200.05,9.2,121.0,2.8,12,0,8,2,200.007,200.007,0,1,0,0,0,0,0,0,0,Between -11 and 5
-471,Tranexamic acid,157.21,63.3,139.0,-2.0,11,2,3,2,157.11,157.11,0,1,0,0,0,0,0,0,0,Between -11 and 5
-472,Prednisolone,360.4,94.8,724.0,1.6,26,3,5,2,360.194,360.194,0,1,0,7,7,0,0,0,0,Between -11 and 5
-473,Levothyroxine,776.87,92.8,420.0,2.4,24,3,5,5,776.687,776.687,0,1,0,1,1,0,0,0,0,Between -11 and 5
-474,Mannitol,182.17,121.0,105.0,-3.1,12,6,6,5,182.079,182.079,0,1,0,4,4,0,0,0,0,Between -11 and 5
-475,Dexamethasone phosphate,472.4,141.0,973.0,0.1,32,4,9,4,472.166,472.166,0,1,0,8,8,0,0,0,0,Between -11 and 5
-476,Citrulline,175.19,118.0,171.0,-4.3,12,4,4,5,175.096,175.096,0,1,0,1,1,0,0,0,0,Between -11 and 5
-477,Hesperidin,610.6,234.0,940.0,-1.1,43,8,15,7,610.19,610.19,0,1,0,11,11,0,0,0,0,Between -11 and 5
-478,Zinc Citrate,574.3,281.0,211.0,,29,2,14,4,571.791,569.794,0,5,0,0,0,0,0,0,0,Larger than 6
-479,Zinc Acetate,183.5,80.3,25.5,,9,0,4,0,181.956,181.956,0,3,0,0,0,0,0,0,0,Larger than 6
-480,Canrenone,340.5,43.4,719.0,2.7,25,0,3,0,340.204,340.204,0,1,0,6,6,0,0,0,0,Between -11 and 5
-481,Hydrocortisone hemisuccinate,462.5,138.0,908.0,1.8,33,3,8,7,462.225,462.225,0,1,0,7,7,0,0,0,0,Between -11 and 5
-482,Methylprednisolone hemisuccinate,474.5,138.0,981.0,2.2,34,3,8,7,474.225,474.225,0,1,0,8,8,0,0,0,0,Between -11 and 5
-483,Azo rubin S,458.5,170.0,880.0,3.3,31,3,9,4,458.024,458.024,0,1,0,0,0,0,0,0,0,Between -11 and 5
-484,"Zinc, bis(D-gluconato-kappaO1,kappaO2)-, (T-4)-",457.7,277.0,385.0,,27,12,14,10,456.046,456.046,0,3,0,8,8,0,0,0,0,Larger than 6
-485,2-acetamido-2-deoxy-beta-D-glucopyranose,221.21,119.0,235.0,-1.7,15,5,6,2,221.09,221.09,0,1,0,5,5,0,0,0,0,Between -11 and 5
-486,Zinc Sulfate,161.4,88.6,62.2,,6,0,4,0,159.881,159.881,0,2,0,0,0,0,0,0,0,Larger than 6
-487,Sodium Thiosulfate,158.11,104.0,82.6,,7,0,4,0,157.908,157.908,0,3,0,0,0,0,0,0,0,Larger than 6
-488,Diacerein,368.3,124.0,683.0,1.9,27,1,8,5,368.053,368.053,0,1,0,0,0,0,0,0,0,Between -11 and 5
-489,Levamisole,204.29,40.9,246.0,1.8,14,0,2,1,204.072,204.072,0,1,0,1,1,0,0,0,0,Between -11 and 5
-490,Cromolyn sodium,512.299,172.0,824.0,,36,1,11,6,512.033,512.033,0,3,0,0,0,0,0,0,0,Larger than 6
-491,Calcium dobesilate,418.4,212.0,228.0,,25,4,10,0,417.934,417.934,0,3,0,0,0,0,0,0,0,Larger than 6
-492,Amoxicillin,365.4,158.0,590.0,-2.0,25,4,7,4,365.105,365.105,0,1,0,4,4,0,0,0,0,Between -11 and 5
-493,Tramadol,263.37,32.7,282.0,2.6,19,1,3,4,263.189,263.189,0,1,0,2,2,0,0,0,0,Between -11 and 5
-494,Almitrine,477.6,69.2,602.0,5.6,35,2,9,10,477.245,477.245,0,1,0,0,0,0,0,0,0,Larger than 6
-495,Pirfenidone,185.22,20.3,285.0,1.9,14,0,1,1,185.084,185.084,0,1,0,0,0,0,0,0,0,Between -11 and 5
-496,Piperacillin,517.6,182.0,982.0,0.5,36,3,8,6,517.163,517.163,0,1,0,4,4,0,0,0,0,Between -11 and 5
-497,Captopril,217.29,58.6,244.0,0.3,14,2,4,3,217.077,217.077,0,1,0,2,2,0,0,0,0,Between -11 and 5
-498,Nicorandil,211.17,97.0,228.0,0.8,15,1,5,4,211.059,211.059,0,1,0,0,0,0,0,0,0,Between -11 and 5
-499,Brequinar,375.4,50.2,551.0,5.6,28,1,5,3,375.107,375.107,0,1,0,0,0,0,0,0,0,Larger than 6
-500,Amantadine hydrochloride,187.71,26.0,144.0,,12,2,1,0,187.113,187.113,0,2,0,0,0,0,0,0,0,Larger than 6
-501,Chloroquine Phosphate,515.9,184.0,359.0,,32,7,11,8,515.135,515.135,0,3,0,1,0,1,0,0,0,Larger than 6
-502,Epigallocatechin Gallate,458.4,197.0,667.0,1.2,33,8,11,4,458.085,458.085,0,1,0,2,2,0,0,0,0,Between -11 and 5
-503,Artemisinin,282.33,54.0,452.0,2.8,20,0,5,0,282.147,282.147,0,1,0,7,7,0,0,0,0,Between -11 and 5
-504,"1-((2S,3R,4S,5S)-3-Fluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione",260.22,99.1,413.0,-0.9,18,3,6,2,260.081,260.081,0,1,0,4,4,0,0,0,0,Between -11 and 5
-505,Bis (2-Carboxyethylgermanium)sesquioxide,339.4,118.0,259.0,,15,2,7,8,339.886,341.885,0,1,0,0,0,0,0,0,0,Larger than 6
-506,Clarithromycin,748.0,183.0,1190.0,3.2,52,4,14,8,747.477,747.477,0,1,0,18,18,0,0,0,0,Between -11 and 5
-507,"2,6-Difluorophenol",130.09,20.2,87.1,2.0,9,1,3,0,130.023,130.023,0,1,0,0,0,0,0,0,0,Between -11 and 5
-508,Moxifloxacin Hydrochloride,437.9,82.1,727.0,,30,3,8,4,437.152,437.152,0,2,0,2,2,0,0,0,0,Larger than 6
-509,Levofloxacin,361.4,73.3,634.0,-0.4,26,1,8,2,361.144,361.144,0,1,0,1,1,0,0,0,0,Between -11 and 5
-510,Olmesartan,446.5,130.0,656.0,3.2,33,3,7,8,446.207,446.207,0,1,0,0,0,0,0,0,0,Between -11 and 5
-511,Tetrasul sulfoxide,340.0,36.3,307.0,5.2,18,0,2,2,339.886,337.889,0,1,0,1,0,1,0,0,0,Larger than 6
-512,Ramelteon,259.339,38.3,331.0,2.7,19,1,2,4,259.157,259.157,0,1,0,1,1,0,0,0,0,Between -11 and 5
-513,Darunavir,547.7,149.0,853.0,2.9,38,3,9,12,547.235,547.235,0,1,0,5,5,0,0,0,0,Between -11 and 5
-514,Galactomannan,504.4,269.0,641.0,-6.3,34,11,16,7,504.169,504.169,0,1,0,15,15,0,0,0,0,Between -11 and 5
-515,Mometasone furoate,521.4,93.8,1020.0,3.9,35,1,6,5,520.142,520.142,0,1,0,8,8,0,0,0,0,Between -11 and 5
-516,Rosuvastatin,481.5,149.0,767.0,1.6,33,3,10,10,481.168,481.168,0,1,0,2,2,0,1,1,0,Between -11 and 5
-517,Tenofovir,287.21,136.0,354.0,-1.6,19,3,8,5,287.078,287.078,0,1,0,1,1,0,0,0,0,Between -11 and 5
-518,3-Cyclopentyl-1-(piperazin-1-yl)propan-1-one,210.32,32.299,206.0,1.6,15,1,2,3,210.173,210.173,0,1,0,0,0,0,0,0,0,Between -11 and 5
-519,Azithromycin Dihydrate,785.0,182.0,1150.0,,54,7,16,7,784.53,784.53,0,3,0,18,18,0,0,0,0,Larger than 6
-520,"D-Alanine, N-methylglycyl-L-arginyl-L-valyl-L-tyrosyl-L-isoleucyl-L-histidyl-L-prolyl-",926.1,358.0,1690.0,-2.3,66,12,13,26,925.513,925.513,0,1,0,8,8,0,0,0,0,Between -11 and 5
-521,Arformoterol,344.4,90.8,388.0,1.8,25,4,5,8,344.174,344.174,0,1,0,2,2,0,0,0,0,Between -11 and 5
-522,3-Hydroxybutyrate,103.1,60.4,63.8,0.1,7,1,3,1,103.04,103.04,-1,1,0,1,0,1,0,0,0,Between -11 and 5
-523,Sitagliptin,407.31,77.0,566.0,0.7,28,1,10,4,407.118,407.118,0,1,0,1,1,0,0,0,0,Between -11 and 5
-524,Best,572.3,101.0,412.0,,18,0,4,0,573.618,575.617,0,1,0,0,0,0,0,0,0,Larger than 6
-525,Eprosartan,424.5,121.0,618.0,4.5,30,2,6,10,424.146,424.146,0,1,0,0,0,0,1,1,0,Between -11 and 5
-526,Isotretinoin,300.4,37.3,567.0,6.3,22,1,2,5,300.209,300.209,0,1,0,0,0,0,4,4,0,Larger than 6
-527,Epoprostenol,352.5,87.0,485.0,2.9,25,3,5,10,352.225,352.225,0,1,0,5,5,0,2,2,0,Between -11 and 5
-528,Camostat mesylate,494.5,200.0,695.0,,34,3,9,9,494.147,494.147,0,2,0,0,0,0,0,0,0,Larger than 6
-529,Fluticasone,444.5,99.9,861.0,3.2,30,2,8,3,444.158,444.158,0,1,0,9,9,0,0,0,0,Between -11 and 5
-530,Tenofovir disoproxil,519.4,185.0,698.0,1.6,35,1,14,17,519.173,519.173,0,1,0,1,1,0,0,0,0,Between -11 and 5
-531,Refanalin,176.24,56.9,170.0,2.2,12,1,2,2,176.041,176.041,0,1,0,0,0,0,1,1,0,Between -11 and 5
-532,Sulodexide,295.29,115.0,363.0,0.2,21,3,8,4,295.128,295.128,0,1,0,4,4,0,0,0,0,Between -11 and 5
-533,Metampicillin,361.4,124.0,603.0,3.0,25,2,6,5,361.11,361.11,0,1,0,4,4,0,0,0,0,Between -11 and 5
-534,Ciclesonide,540.7,99.1,1100.0,5.3,39,1,7,6,540.309,540.309,0,1,0,9,9,0,0,0,0,Larger than 6
-535,Dutasteride,528.5,58.2,964.0,5.4,37,2,8,2,528.221,528.221,0,1,0,7,7,0,0,0,0,Larger than 6
-536,Prasugrel,373.4,74.8,555.0,3.6,26,0,6,6,373.115,373.115,0,1,0,1,0,1,0,0,0,Between -11 and 5
-537,Almitrine mesylate,669.8,195.0,694.0,,45,4,15,10,669.221,669.221,0,3,0,0,0,0,0,0,0,Larger than 6
-538,Ile-Ser,218.25,113.0,232.0,-3.3,15,4,5,6,218.127,218.127,0,1,0,3,3,0,0,0,0,Between -11 and 5
-539,butyl (3-(4-((1H-imidazol-1-yl)methyl)phenyl)-5-isobutylthiophen-2-yl)sulfonylcarbamate,475.6,127.0,690.0,5.3,32,1,6,11,475.16,475.16,0,1,0,0,0,0,0,0,0,Larger than 6
-540,Vidofludimus,355.4,75.6,576.0,3.4,26,2,5,5,355.122,355.122,0,1,0,0,0,0,0,0,0,Between -11 and 5
-541,Reparixin,283.39,71.6,389.0,2.9,19,1,3,5,283.124,283.124,0,1,0,1,1,0,0,0,0,Between -11 and 5
-542,Ticagrelor,522.6,164.0,736.0,2.0,36,4,12,10,522.186,522.186,0,1,0,6,6,0,0,0,0,Between -11 and 5
-543,Anhydrous Ceftriaxone Sodium,577.6,288.0,1110.0,,37,4,13,8,577.036,577.036,0,2,0,2,2,0,1,1,0,Larger than 6
-544,Tradipitant,587.9,73.6,865.0,6.2,41,0,11,6,587.095,587.095,0,1,0,0,0,0,0,0,0,Larger than 6
-545,CID 9939931,701.6,316.0,819.0,,47,12,9,18,700.261,700.261,0,2,0,4,4,0,2,2,0,Larger than 6
-546,1-Palmitoyl-2-linoleoyl-3-acetyl-rac-glycerol,635.0,78.9,744.0,14.1,45,0,6,36,634.517,634.517,0,1,0,1,0,1,2,2,0,Larger than 6
-547,Zinforo,744.7,368.0,1240.0,,47,5,19,10,744.031,744.031,0,2,0,2,2,0,1,1,0,Larger than 6
-548,Linagliptin,472.5,114.0,885.0,1.9,35,1,7,4,472.234,472.234,0,1,0,1,1,0,0,0,0,Between -11 and 5
-549,"2-(2-Chloro-4-iodophenylamino)-3,4-difluorobenzoic acid",409.55,49.3,363.0,5.0,20,2,5,3,408.918,408.918,0,1,0,0,0,0,0,0,0,Between -11 and 5
-550,Edoxaban,548.1,165.0,880.0,1.4,37,3,8,5,547.177,547.177,0,1,0,3,3,0,0,0,0,Between -11 and 5
-551,Transcrocetinate sodium,372.4,80.3,597.0,,26,0,4,6,372.131,372.131,0,3,0,0,0,0,7,7,0,Larger than 6
-552,Galidesivir,265.27,140.0,334.0,-2.1,19,6,7,2,265.117,265.117,0,1,0,4,4,0,0,0,0,Between -11 and 5
-553,"4,5-Dihydro-3-phenyl-5-isoxazoleacetic acid",205.21,58.9,269.0,1.3,15,1,4,3,205.074,205.074,0,1,0,1,0,1,0,0,0,Between -11 and 5
-554,5-Cholesten-3beta-25-diol-3-sulfate,482.7,92.2,858.0,6.4,33,2,5,7,482.307,482.307,0,1,0,8,8,0,0,0,0,Larger than 6
-555,"disodium;(6R,7R)-7-[[(2Z)-2-(2-amino-1,3-thiazol-4-yl)-2-methoxyiminoacetyl]amino]-3-[(2-methyl-6-oxido-5-oxo-1,2,4-triazin-3-yl)sulfanylmethyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate;hydrate",616.6,298.0,1120.0,,39,3,15,7,616.021,616.021,0,4,0,2,2,0,1,1,0,Larger than 6
-556,Losartan potassium,461.0,77.7,526.0,,31,1,6,8,460.118,460.118,0,2,0,0,0,0,0,0,0,Larger than 6
-557,Lopinavir and ritonavir,1349.7,322.0,1980.0,,96,8,14,33,1348.68,1348.68,0,2,0,8,8,0,0,0,0,Larger than 6
-558,"N-[5-[[2-(2,6-dimethylphenoxy)acetyl]amino]-4-hydroxy-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxo-1,3-diazinan-1-yl)butanamide;1,3-thiazol-5-ylmethyl N-[(2S,3S,5S)-3-hydroxy-5-[[(2S)-3-methyl-2-[[methyl-[(2-propan-2-yl-1,3-thiazol-4-yl)methyl]carbamoyl]amino]butanoyl]amino]-1,6-diphenylhexan-2-yl]carbamate",1349.7,322.0,1980.0,,96,8,14,33,1348.68,1348.68,0,2,0,8,4,4,0,0,0,Larger than 6
-559,"S-[2-[3-[[(2R)-4-[[[(2R,3S,4R,5R)-5-(6-aminopurin-9-yl)-4-hydroxy-3-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl]oxy-2-hydroxy-3,3-dimethylbutanoyl]amino]propanoylamino]ethyl] (2S)-2-[4-(2-methylpropyl)phenyl]propanethioate",955.8,389.0,1660.0,-1.9,62,9,22,24,955.235,955.235,0,1,0,6,6,0,0,0,0,Between -11 and 5
-560,Bivalirudin,2180.3,902.0,4950.0,-7.1,155,28,35,67,2179.99,2178.99,0,1,0,16,16,0,0,0,0,Between -11 and 5
-561,Thymalfasin,3108.3,1460.0,7190.0,-24.0,217,49,59,111,3107.51,3106.5,0,1,0,32,32,0,0,0,0,Smaller than -10
-562,CID 16219160,397.6,74.6,707.0,,27,2,4,3,397.178,397.178,0,2,0,6,6,0,0,0,0,Larger than 6
-563,Sodium valproate,166.19,40.1,98.3,,11,0,2,5,166.097,166.097,0,2,0,0,0,0,0,0,0,Larger than 6
-564,"N,N-Diethyl-(2-(4-(2-(18F)fluoroethoxy)phenyl)-5,7-dimethylpyrazolo(1,5-A)pyrimidine-3-YL)acetamide",397.5,59.7,525.0,3.2,29,0,5,8,397.214,397.214,0,1,1,0,0,0,0,0,0,Between -11 and 5
-565,Amoxicillin sodium,387.4,161.0,596.0,,26,3,7,4,387.086,387.086,0,2,0,4,4,0,0,0,0,Larger than 6
-566,Potassium canrenoate,396.6,77.4,713.0,,27,1,4,3,396.17,396.17,0,2,0,6,6,0,0,0,0,Larger than 6
-567,Hydrocortisone sodium succinate,484.5,141.0,915.0,,34,2,8,7,484.207,484.207,0,2,0,7,7,0,0,0,0,Larger than 6
-568,Piperacillin-tazobactam,839.8,315.0,1560.0,,57,3,15,9,839.198,839.198,0,3,0,7,7,0,0,0,0,Larger than 6
-569,Antroquinonol,390.6,55.8,648.0,5.8,28,1,4,10,390.277,390.277,0,1,0,3,3,0,2,2,0,Larger than 6
-570,Brilacidin,936.9,314.0,1560.0,0.3,66,10,18,20,936.394,936.394,0,1,0,2,2,0,0,0,0,Between -11 and 5
-571,Ruxolitinib phosphate,404.4,161.0,503.0,,28,4,8,4,404.136,404.136,0,2,0,1,1,0,0,0,0,Larger than 6
-572,Baricitinib,371.4,129.0,678.0,-0.5,26,1,7,5,371.116,371.116,0,1,0,0,0,0,0,0,0,Between -11 and 5
-573,"(2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile",291.26,150.0,456.0,-1.4,21,4,8,2,291.097,291.097,0,1,0,4,4,0,0,0,0,Between -11 and 5
-574,Metformin glycinate,204.23,155.0,175.0,,14,5,4,3,204.133,204.133,0,2,0,0,0,0,0,0,0,Larger than 6
-575,1-Palmityl-2-(4-carboxybutyl)-SN-glycero-3-phosphocholine,581.8,114.0,606.0,6.7,39,1,8,30,581.406,581.406,0,1,0,1,1,0,0,0,0,Larger than 6
-576,3-phenyl-4-propyl-1-(pyridin-2-yl)-1H-pyrazol-5-ol,279.34,45.2,412.0,3.7,21,1,3,4,279.137,279.137,0,1,0,0,0,0,0,0,0,Between -11 and 5
-577,Ozanimod,404.5,104.0,609.0,3.1,30,2,7,7,404.185,404.185,0,1,0,1,1,0,0,0,0,Between -11 and 5
-578,Sabizabulin,377.4,89.2,534.0,3.4,28,2,5,6,377.138,377.138,0,1,0,0,0,0,0,0,0,Between -11 and 5
-579,Zavegepant,638.8,117.0,1160.0,3.1,47,3,6,6,638.369,638.369,0,1,0,1,1,0,0,0,0,Between -11 and 5
-580,CID 53477736,749.0,180.0,1150.0,4.0,52,5,14,7,748.509,748.509,0,1,0,18,18,0,0,0,0,Between -11 and 5
-581,Danoprevir (RG7227),731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,5,0,1,1,0,Between -11 and 5
-582,Solu-Medrol,497.5,138.0,981.0,,35,3,8,7,497.215,497.215,1,2,0,8,8,0,0,0,0,Larger than 6
-583,Vericiguat,426.4,147.0,622.0,1.5,31,3,10,5,426.136,426.136,0,1,0,0,0,0,0,0,0,Between -11 and 5
-584,Aldose reductase-IN-1,421.4,137.0,706.0,2.3,29,1,11,4,421.046,421.046,0,1,0,0,0,0,0,0,0,Between -11 and 5
-585,Ixazomib citrate,517.1,168.0,797.0,,34,4,9,11,516.087,516.087,0,1,0,1,1,0,0,0,0,Larger than 6
-586,Vidofludimus calcium anhydrous,748.8,157.0,571.0,,53,2,10,8,748.191,748.191,0,3,0,0,0,0,0,0,0,Larger than 6
-587,Upadacitinib,380.4,78.3,561.0,2.7,27,2,6,3,380.157,380.157,0,1,0,2,2,0,0,0,0,Between -11 and 5
-588,Asapiprant,501.6,131.0,789.0,3.1,35,1,10,9,501.157,501.157,0,1,0,0,0,0,0,0,0,Between -11 and 5
-589,"1,1'-hexamethylene bis[5-(p-chlorophenyl) biguanide] di-D-gluconate",735.7,317.0,819.0,,48,13,10,18,734.249,734.249,0,3,0,4,4,0,2,2,0,Larger than 6
-590,Proxalutamide,517.5,118.0,894.0,4.3,36,0,10,6,517.12,517.12,0,1,0,0,0,0,0,0,0,Between -11 and 5
-591,Rocefin,652.6,300.0,1120.0,,41,5,17,7,652.042,652.042,0,6,0,2,2,0,1,1,0,Larger than 6
-592,Galidesivir hydrochloride,301.73,140.0,334.0,,20,7,7,2,301.094,301.094,0,2,0,4,4,0,0,0,0,Larger than 6
-593,Entresto,1916.0,396.0,1140.0,,135,7,29,40,1915.81,1914.81,0,15,0,6,6,0,0,0,0,Larger than 6
-594,"(2R,6S,12Z,13aS,14aR,16aS)-6-[(tert-Butoxycarbonyl)amino]-14a-[(cyclopropylsulfonyl)carbamoyl]-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl 4-fluoro-1,3-dihydro-2H-isoindole-2-carboxylate",731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,5,0,1,1,0,Between -11 and 5
-595,Harvoni,1418.4,327.0,2730.0,,101,7,21,23,1417.58,1417.58,0,2,0,12,12,0,0,0,0,Larger than 6
-596,Abivertinib,487.5,98.4,752.0,4.2,36,3,8,7,487.213,487.213,0,1,0,0,0,0,0,0,0,Between -11 and 5
-597,Legalon SIL,1453.1,495.0,1200.0,,102,6,32,24,1452.23,1452.23,0,6,0,8,8,0,0,0,0,Larger than 6
-598,Maltofer,449.16,200.0,367.0,,27,11,14,8,449.059,449.059,0,5,0,9,9,0,0,0,0,Larger than 6
-599,Zunsemetinib,513.9,101.0,888.0,2.5,36,1,9,6,513.138,513.138,0,1,0,0,0,0,0,0,0,Between -11 and 5
-600,Treamid,318.37,116.0,345.0,-0.7,23,4,4,10,318.18,318.18,0,1,0,0,0,0,0,0,0,Between -11 and 5
-601,CID 87071853,1003.2,299.0,767.0,,67,6,20,20,1002.3,1002.3,0,3,0,4,4,0,2,2,0,Larger than 6
-602,"disodium;[2-[(8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihydroxy-10,13,16-trimethyl-3-oxo-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl]-2-oxoethyl] phosphate;hydrate",534.4,148.0,962.0,,35,3,10,3,534.141,534.141,0,4,0,8,8,0,0,0,0,Larger than 6
-603,Heparin sodium,1157.9,652.0,2410.0,,71,15,38,21,1157.0,1157.0,1,2,0,20,0,20,0,0,0,Larger than 6
-604,CID 101731853,2088.6,933.0,4400.0,3.1,140,40,48,73,2086.96,2086.96,0,1,0,18,16,2,0,0,0,Between -11 and 5
-605,"N-(5-Oxidanyl-1,3-Benzothiazol-2-Yl)ethanamide",208.24,90.5,237.0,1.5,14,2,4,1,208.031,208.031,0,1,0,0,0,0,0,0,0,Between -11 and 5
-606,Danicopan,580.4,123.0,891.0,3.3,38,1,8,6,579.103,579.103,0,1,0,2,2,0,0,0,0,Between -11 and 5
-607,"Disodium;2-[[2-[[4-(2,2-dimethylpropanoyloxy)phenyl]sulfonylamino]benzoyl]amino]acetate",479.4,150.0,726.0,,32,2,8,8,479.086,479.086,1,3,0,0,0,0,0,0,0,Larger than 6
-608,"propan-2-yl (2S)-2-[[[(3R,4R,5R)-5-[2-amino-6-(methylamino)purin-9-yl]-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate",581.5,185.0,919.0,1.7,40,4,14,12,581.216,581.216,0,1,0,6,5,1,0,0,0,Between -11 and 5
-609,"[(1S,4R,6S,7E,18R)-4-(cyclopropylsulfonylcarbamoyl)-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-3,16-diazatricyclo[14.3.0.04,6]nonadec-7-en-18-yl] 4-fluoro-1,3-dihydroisoindole-2-carboxylate",731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,4,1,1,1,0,Between -11 and 5
-610,Eclitasertib,378.4,113.0,570.0,1.7,28,2,6,4,378.144,378.144,0,1,0,1,1,0,0,0,0,Between -11 and 5
-611,Dazcapistat,395.4,115.0,611.0,2.9,29,2,6,7,395.128,395.128,0,1,0,1,0,1,0,0,0,Between -11 and 5
-612,Bexotegrast,492.6,113.0,655.0,1.8,36,3,9,14,492.285,492.285,0,1,0,1,1,0,0,0,0,Between -11 and 5
-613,Estetrol monohydrate,322.4,81.9,441.0,,23,5,5,0,322.178,322.178,0,2,0,7,7,0,0,0,0,Larger than 6
-614,Sildenafil,474.6,118.0,838.0,1.5,33,1,8,7,474.205,474.205,0,1,0,0,0,0,0,0,0,Between -11 and 5
-615,Azilsartan,456.4,115.0,783.0,4.4,34,2,7,7,456.143,456.143,0,1,0,0,0,0,0,0,0,Between -11 and 5
-616,Echinochrome A,266.2,135.0,455.0,2.0,19,5,7,1,266.043,266.043,0,1,0,0,0,0,0,0,0,Between -11 and 5
-617,F-Arag F-18,284.23,135.0,449.0,-0.9,20,4,7,2,284.09,284.09,0,1,1,4,4,0,0,0,0,Between -11 and 5
-618,Apabetalone,370.4,89.4,543.0,2.3,27,2,6,6,370.153,370.153,0,1,0,0,0,0,0,0,0,Between -11 and 5
-619,"4-acetamidobenzoic acid;9-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1H-purin-6-one;(2R)-1-(dimethylamino)propan-2-ol",1115.2,399.0,658.0,,79,13,22,14,1114.55,1114.55,0,7,0,7,7,0,0,0,0,Larger than 6
-620,"[(1R,3R)-4-[(15Z,17E)-16-formyl-18-(4-hydroxy-2,2,6,6-tetramethylcyclohexyl)-3,7,12-trimethyl-14-oxooctadeca-2,4,6,8,10,12,15,17-octaenylidene]-3-hydroxy-3,5,5-trimethylcyclohexyl] acetate",672.9,101.0,1480.0,9.3,49,2,6,13,672.439,672.439,0,1,0,2,2,0,9,2,7,Larger than 6
-621,Abivertinib maleate,639.6,175.0,871.0,,46,7,14,9,639.245,639.245,0,4,0,0,0,0,1,1,0,Larger than 6
-622,"7-[[2-(2-Amino-1,3-thiazol-4-yl)-2-(2,2-dimethylpropanoyloxymethoxyimino)acetyl]amino]-3-ethenyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid",509.6,227.0,961.0,1.1,34,3,12,10,509.104,509.104,0,1,0,2,0,2,1,0,1,Between -11 and 5
-623,Unii-T5UX5skk2S,452.5,120.0,723.0,3.5,33,4,4,9,452.242,452.242,0,1,0,3,3,0,0,0,0,Between -11 and 5
-624,"azane;(2R,3S,4S,5R,6R)-2-(hydroxymethyl)-6-[(2R,3S,4R,5R,6S)-4,5,6-trihydroxy-2-(hydroxymethyl)oxan-3-yl]oxyoxane-3,4,5-triol",359.33,191.0,382.0,,24,9,12,4,359.143,359.143,0,2,0,10,10,0,0,0,0,Larger than 6
-625,Nezulcitinib,527.7,104.0,866.0,3.6,39,3,6,6,527.301,527.301,0,1,0,1,1,0,0,0,0,Between -11 and 5
-626,P9Zqs28F8C,403.5,86.9,712.0,3.6,28,2,4,4,403.193,403.193,0,1,0,1,1,0,1,1,0,Between -11 and 5
-627,Lufotrelvir,552.5,196.0,927.0,0.5,38,6,9,13,552.199,552.199,0,1,0,3,3,0,0,0,0,Between -11 and 5
-628,Nirmatrelvir,499.5,131.0,964.0,2.2,35,3,8,7,499.241,499.241,0,1,0,6,6,0,0,0,0,Between -11 and 5
-629,"hexasodium;4-[[(2S,4R)-5-ethoxy-4-methyl-5-oxo-1-(4-phenylphenyl)pentan-2-yl]amino]-4-oxobutanoate;hydride;(2S)-3-methyl-2-[pentanoyl-[[4-[2-(1,2,3-triaza-4-azanidacyclopenta-2,5-dien-5-yl)phenyl]phenyl]methyl]amino]butanoate;pentahydrate",1922.0,396.0,1140.0,,135,7,35,40,1921.86,1920.85,-6,21,0,6,6,0,0,0,0,Larger than 6
-630,Nangibotide,1342.5,634.0,2630.0,-8.3,92,20,24,45,1341.53,1341.53,0,1,0,10,10,0,0,0,0,Between -11 and 5
-
-
-
-
-
-
- ]]>
- Python
- Shiny
- Pandas
- Plotly
- PubChem
- Cheminformatics
- https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_app_embed.html
- Sun, 07 May 2023 12:00:00 GMT
-
-
- Shinylive app in Python
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_data_prep.html
-
-
Brief introduction
-
Since I’ve had a lot of fun building a Shiny app in R last time, I was on track to build another Shiny app again but using Python instead. So here in this post, I’ll talk about the data wrangling process to prepare the final dataset needed to build a Shinylive app in Python. The actual Shinylive app deployment and access will be shown in a separate post after this one.
-
-
-
-
Source of data
-
The dataset used for this Shiny app in Python was from PubChem (link here). There were a total of 631 compounds at the time when I downloaded them as .csv file, along with their relevant compound data. I only picked this dataset randomly, as the focus would be more on app building, but it was nice to see an interactive web app being built and used for a domain such as pharmaceutical research.
-
-
-
-
Import Polars
-
Polars dataframe library was used again this time.
-
-
import polars as pl
-
-
-
-
-
Reading .csv file
-
-
pc = pl.read_csv("pubchem.csv")
-pc.head()
-
-
-
-
-shape: (5, 38)
-
-
-
-
-cid
-
-
-cmpdname
-
-
-cmpdsynonym
-
-
-mw
-
-
-mf
-
-
-polararea
-
-
-complexity
-
-
-xlogp
-
-
-heavycnt
-
-
-hbonddonor
-
-
-hbondacc
-
-
-rotbonds
-
-
-inchi
-
-
-isosmiles
-
-
-canonicalsmiles
-
-
-inchikey
-
-
-iupacname
-
-
-exactmass
-
-
-monoisotopicmass
-
-
-charge
-
-
-covalentunitcnt
-
-
-isotopeatomcnt
-
-
-totalatomstereocnt
-
-
-definedatomstereocnt
-
-
-undefinedatomstereocnt
-
-
-totalbondstereocnt
-
-
-definedbondstereocnt
-
-
-undefinedbondstereocnt
-
-
-pclidcnt
-
-
-gpidcnt
-
-
-meshheadings
-
-
-annothits
-
-
-annothitcnt
-
-
-aids
-
-
-cidcdate
-
-
-sidsrcname
-
-
-depcatg
-
-
-annotation
-
-
-
-
-i64
-
-
-str
-
-
-str
-
-
-f64
-
-
-str
-
-
-f64
-
-
-f64
-
-
-str
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-f64
-
-
-f64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-str
-
-
-str
-
-
-i64
-
-
-str
-
-
-i64
-
-
-str
-
-
-str
-
-
-str
-
-
-
-
-
-
-5280453
-
-
-"Calcitriol"
-
-
-"calcitriol|322...
-
-
-416.6
-
-
-"C27H44O3"
-
-
-60.7
-
-
-688.0
-
-
-"5.100"
-
-
-30
-
-
-3
-
-
-3
-
-
-6
-
-
-"InChI=1S/C27H4...
-
-
-"C[C@H](CCCC(C)...
-
-
-"CC(CCCC(C)(C)O...
-
-
-"GMRQFYUYWCNGIN...
-
-
-"(1R,3S,5Z)-5-[...
-
-
-416.329
-
-
-416.329
-
-
-0
-
-
-1
-
-
-0
-
-
-6
-
-
-6
-
-
-0
-
-
-2
-
-
-2
-
-
-0
-
-
-22311
-
-
-46029
-
-
-"Calcitriol"
-
-
-"Biological Tes...
-
-
-12
-
-
-"485|631|731|78...
-
-
-20040916
-
-
-"A2B Chem|AA BL...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-9962735
-
-
-"Ubiquinol"
-
-
-"ubiquinol|992-...
-
-
-865.4
-
-
-"C59H92O4"
-
-
-58.9
-
-
-1600.0
-
-
-"20.200"
-
-
-63
-
-
-2
-
-
-4
-
-
-31
-
-
-"InChI=1S/C59H9...
-
-
-"CC1=C(C(=C(C(=...
-
-
-"CC1=C(C(=C(C(=...
-
-
-"QNTNKSLOFHEFPK...
-
-
-"2-[(2E,6E,10E,...
-
-
-864.7
-
-
-864.7
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-9
-
-
-9
-
-
-0
-
-
-2732
-
-
-21358
-
-
-"NULL"
-
-
-"Chemical and P...
-
-
-7
-
-
-"NULL"
-
-
-20061025
-
-
-"001Chemical|A2...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-5961
-
-
-"Glutamine"
-
-
-"L-glutamine|gl...
-
-
-146.14
-
-
-"C5H10N2O3"
-
-
-106.0
-
-
-146.0
-
-
-"-3.100"
-
-
-10
-
-
-3
-
-
-4
-
-
-4
-
-
-"InChI=1S/C5H10...
-
-
-"C(CC(=O)N)[C@@...
-
-
-"C(CC(=O)N)C(C(...
-
-
-"ZDXPYRJPNDTMRX...
-
-
-"(2S)-2,5-diami...
-
-
-146.069
-
-
-146.069
-
-
-0
-
-
-1
-
-
-0
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-88218
-
-
-399
-
-
-"Glutamine"
-
-
-"Biological Tes...
-
-
-12
-
-
-"422|429|436|54...
-
-
-20040916
-
-
-"001Chemical|3B...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-2244
-
-
-"Aspirin"
-
-
-"aspirin|ACETYL...
-
-
-180.16
-
-
-"C9H8O4"
-
-
-63.6
-
-
-212.0
-
-
-"1.200"
-
-
-13
-
-
-1
-
-
-4
-
-
-3
-
-
-"InChI=1S/C9H8O...
-
-
-"CC(=O)OC1=CC=C...
-
-
-"CC(=O)OC1=CC=C...
-
-
-"BSYNRYMUTXBXSQ...
-
-
-"2-acetyloxyben...
-
-
-180.042
-
-
-180.042
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-127012
-
-
-364455
-
-
-"Aspirin"
-
-
-"Biological Tes...
-
-
-12
-
-
-"1|3|9|15|19|21...
-
-
-20040916
-
-
-"001Chemical|3B...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-457
-
-
-"1-Methylnicoti...
-
-
-"1-methylnicoti...
-
-
-137.16
-
-
-"C7H9N2O+"
-
-
-47.0
-
-
-136.0
-
-
-"-0.100"
-
-
-10
-
-
-1
-
-
-1
-
-
-1
-
-
-"InChI=1S/C7H8N...
-
-
-"C[N+]1=CC=CC(=...
-
-
-"C[N+]1=CC=CC(=...
-
-
-"LDHMAVIPBRSVRG...
-
-
-"1-methylpyridi...
-
-
-137.071
-
-
-137.071
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-310
-
-
-674
-
-
-"NULL"
-
-
-"Biological Tes...
-
-
-8
-
-
-"61001|61002|14...
-
-
-20040916
-
-
-"001Chemical|3B...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-
-
-
-
-
-
-
Quick look at the data
-
I decided to comment out the code below to keep the post at a reasonable length for reading purpose, but they were very handy for a quick glimpse of the data content.
-
-
# Quick overview of the variables in each column in the dataset
-# Uncomment line below if needed to run
-#print(pc.glimpse())
-
-# Quick look at all column names
-# Uncomment line below if needed to run
-#pc.columns
-
-
-
-
-
Check for nulls in dataset
-
-
pc.null_count()
-
-
-
-
-shape: (1, 38)
-
-
-
-
-cid
-
-
-cmpdname
-
-
-cmpdsynonym
-
-
-mw
-
-
-mf
-
-
-polararea
-
-
-complexity
-
-
-xlogp
-
-
-heavycnt
-
-
-hbonddonor
-
-
-hbondacc
-
-
-rotbonds
-
-
-inchi
-
-
-isosmiles
-
-
-canonicalsmiles
-
-
-inchikey
-
-
-iupacname
-
-
-exactmass
-
-
-monoisotopicmass
-
-
-charge
-
-
-covalentunitcnt
-
-
-isotopeatomcnt
-
-
-totalatomstereocnt
-
-
-definedatomstereocnt
-
-
-undefinedatomstereocnt
-
-
-totalbondstereocnt
-
-
-definedbondstereocnt
-
-
-undefinedbondstereocnt
-
-
-pclidcnt
-
-
-gpidcnt
-
-
-meshheadings
-
-
-annothits
-
-
-annothitcnt
-
-
-aids
-
-
-cidcdate
-
-
-sidsrcname
-
-
-depcatg
-
-
-annotation
-
-
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-u32
-
-
-
-
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-
-
-
-
-
-
-
-
-
Change column names as needed
-
-
# Change column names
-pc_cov = pc.rename(
- {
-"cmpdname": "Compound name",
-"cmpdsynonym": "Synonyms",
-"mw": "Molecular weight",
-"mf": "Molecular formula",
-"polararea": "Polar surface area",
-"complexity": "Complexity",
-"xlogp": "Partition coefficients",
-"heavycnt": "Heavy atom count",
-"hbonddonor": "Hydrogen bond donor count",
-"hbondacc": "Hydrogen bond acceptor count",
-"rotbonds": "Rotatable bond count",
-"exactmass": "Exact mass",
-"monoisotopicmass": "Monoisotopic mass",
-"charge": "Formal charge",
-"covalentunitcnt": "Covalently-bonded unit count",
-"isotopeatomcnt": "Isotope atom count",
-"totalatomstereocnt": "Total atom stereocenter count",
-"definedatomstereocnt": "Defined atom stereocenter count",
-"undefinedatomstereocnt": "Undefined atoms stereocenter count",
-"totalbondstereocnt": "Total bond stereocenter count",
-"definedbondstereocnt": "Defined bond stereocenter count",
-"undefinedbondstereocnt": "Undefined bond stereocenter count",
-"meshheadings": "MeSH headings"
- }
-)
-
-pc_cov.head()
-
-
-
-
-shape: (5, 38)
-
-
-
-
-cid
-
-
-Compound name
-
-
-Synonyms
-
-
-Molecular weight
-
-
-Molecular formula
-
-
-Polar surface area
-
-
-Complexity
-
-
-Partition coefficients
-
-
-Heavy atom count
-
-
-Hydrogen bond donor count
-
-
-Hydrogen bond acceptor count
-
-
-Rotatable bond count
-
-
-inchi
-
-
-isosmiles
-
-
-canonicalsmiles
-
-
-inchikey
-
-
-iupacname
-
-
-Exact mass
-
-
-Monoisotopic mass
-
-
-Formal charge
-
-
-Covalently-bonded unit count
-
-
-Isotope atom count
-
-
-Total atom stereocenter count
-
-
-Defined atom stereocenter count
-
-
-Undefined atoms stereocenter count
-
-
-Total bond stereocenter count
-
-
-Defined bond stereocenter count
-
-
-Undefined bond stereocenter count
-
-
-pclidcnt
-
-
-gpidcnt
-
-
-MeSH headings
-
-
-annothits
-
-
-annothitcnt
-
-
-aids
-
-
-cidcdate
-
-
-sidsrcname
-
-
-depcatg
-
-
-annotation
-
-
-
-
-i64
-
-
-str
-
-
-str
-
-
-f64
-
-
-str
-
-
-f64
-
-
-f64
-
-
-str
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-f64
-
-
-f64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-str
-
-
-str
-
-
-i64
-
-
-str
-
-
-i64
-
-
-str
-
-
-str
-
-
-str
-
-
-
-
-
-
-5280453
-
-
-"Calcitriol"
-
-
-"calcitriol|322...
-
-
-416.6
-
-
-"C27H44O3"
-
-
-60.7
-
-
-688.0
-
-
-"5.100"
-
-
-30
-
-
-3
-
-
-3
-
-
-6
-
-
-"InChI=1S/C27H4...
-
-
-"C[C@H](CCCC(C)...
-
-
-"CC(CCCC(C)(C)O...
-
-
-"GMRQFYUYWCNGIN...
-
-
-"(1R,3S,5Z)-5-[...
-
-
-416.329
-
-
-416.329
-
-
-0
-
-
-1
-
-
-0
-
-
-6
-
-
-6
-
-
-0
-
-
-2
-
-
-2
-
-
-0
-
-
-22311
-
-
-46029
-
-
-"Calcitriol"
-
-
-"Biological Tes...
-
-
-12
-
-
-"485|631|731|78...
-
-
-20040916
-
-
-"A2B Chem|AA BL...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-9962735
-
-
-"Ubiquinol"
-
-
-"ubiquinol|992-...
-
-
-865.4
-
-
-"C59H92O4"
-
-
-58.9
-
-
-1600.0
-
-
-"20.200"
-
-
-63
-
-
-2
-
-
-4
-
-
-31
-
-
-"InChI=1S/C59H9...
-
-
-"CC1=C(C(=C(C(=...
-
-
-"CC1=C(C(=C(C(=...
-
-
-"QNTNKSLOFHEFPK...
-
-
-"2-[(2E,6E,10E,...
-
-
-864.7
-
-
-864.7
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-9
-
-
-9
-
-
-0
-
-
-2732
-
-
-21358
-
-
-"NULL"
-
-
-"Chemical and P...
-
-
-7
-
-
-"NULL"
-
-
-20061025
-
-
-"001Chemical|A2...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-5961
-
-
-"Glutamine"
-
-
-"L-glutamine|gl...
-
-
-146.14
-
-
-"C5H10N2O3"
-
-
-106.0
-
-
-146.0
-
-
-"-3.100"
-
-
-10
-
-
-3
-
-
-4
-
-
-4
-
-
-"InChI=1S/C5H10...
-
-
-"C(CC(=O)N)[C@@...
-
-
-"C(CC(=O)N)C(C(...
-
-
-"ZDXPYRJPNDTMRX...
-
-
-"(2S)-2,5-diami...
-
-
-146.069
-
-
-146.069
-
-
-0
-
-
-1
-
-
-0
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-88218
-
-
-399
-
-
-"Glutamine"
-
-
-"Biological Tes...
-
-
-12
-
-
-"422|429|436|54...
-
-
-20040916
-
-
-"001Chemical|3B...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-2244
-
-
-"Aspirin"
-
-
-"aspirin|ACETYL...
-
-
-180.16
-
-
-"C9H8O4"
-
-
-63.6
-
-
-212.0
-
-
-"1.200"
-
-
-13
-
-
-1
-
-
-4
-
-
-3
-
-
-"InChI=1S/C9H8O...
-
-
-"CC(=O)OC1=CC=C...
-
-
-"CC(=O)OC1=CC=C...
-
-
-"BSYNRYMUTXBXSQ...
-
-
-"2-acetyloxyben...
-
-
-180.042
-
-
-180.042
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-127012
-
-
-364455
-
-
-"Aspirin"
-
-
-"Biological Tes...
-
-
-12
-
-
-"1|3|9|15|19|21...
-
-
-20040916
-
-
-"001Chemical|3B...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-457
-
-
-"1-Methylnicoti...
-
-
-"1-methylnicoti...
-
-
-137.16
-
-
-"C7H9N2O+"
-
-
-47.0
-
-
-136.0
-
-
-"-0.100"
-
-
-10
-
-
-1
-
-
-1
-
-
-1
-
-
-"InChI=1S/C7H8N...
-
-
-"C[N+]1=CC=CC(=...
-
-
-"C[N+]1=CC=CC(=...
-
-
-"LDHMAVIPBRSVRG...
-
-
-"1-methylpyridi...
-
-
-137.071
-
-
-137.071
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-310
-
-
-674
-
-
-"NULL"
-
-
-"Biological Tes...
-
-
-8
-
-
-"61001|61002|14...
-
-
-20040916
-
-
-"001Chemical|3B...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-
-
-
-
-
-
-
Definitions of molecular properties in this PubChem dataset
-
The definitions for some of the column names were shown below, which were mainly derived and adapted from PubChem:
-
Note: please refer to PubChem documentations for full definitions
-
-
Molecular weight - molecular mass of compounds measured in daltons
-
Topological polar surface area - measured as an estimate of polar surface area of a molecule (i.e. the surface sum over polar atoms in a molecule), with units in angstrom squared (Å2)
-
Complexity - complexity rating for compounds, based on Bertz/Hendrickson/Ihlenfeldt formula as a rough estimation of how complex a compound was structurally
-
Partition coefficients (xlogp) - predicted octanol-water partition coefficient as a measure of the hydrophilicity or hydrophobicity of a molecule
-
Heavy atom count - number of heavy atoms e.g. non-hydrogen atoms in the compound
-
Hydrogen bond donor count - number of hydrogen bond donors in the compound
-
Hydrogen bond acceptor count - number of hydrogen bond acceptors in the compound
-
Rotatable bond count - defined as any single-order non-ring bond, where atoms on either side of the bond were in turn bound to non-terminal heavy atoms (e.g. non-hydrogen). Rotation around the bond axis would change overall molecule shape and generate conformers which could be distinguished by standard spectroscopic methods
-
Exact mass - exact mass of an isotopic species, obtained by summing masses of individual isotopes of the molecule
-
Monoisotopic mass - sum of the masses of atoms in a molecule, using unbound, ground-state, rest mass of principal (or most abundant) isotope for each element instead of isotopic average mass
-
Formal charge - the difference between the number of valence electrons of each atom, and the number of electrons the atom was associated with, assumed any shared electrons were equally shared between the two bonded atoms
-
Covalently-bonded unit count - a group of atoms connected by covalent bonds, ignoring other bond types (or a single atom without covalent bonds), representing number of such units in the compound
-
Isotope atom count - number of isotopes that were not most abundant for the corresponding chemical elements. Isotopes were variants of a chemical element that differed in neutron number
-
Defined atom stereocenter count - atom stereocenter (or chiral center) was where an atom was attached to 4 different types of atoms or groups of atoms in a tetrahedral arrangement. It could either be (R)- or (S)- configurations. Some of the compounds e.g. racemic mixtures, could have undefined atom stereocenter, where (R/S)-config was not specifically defined. Defined atom stereocenter count was the number of atom stereocenters where configurations were specifically defined
-
Undefined atoms stereocenter count - this was the undefined version of the atoms stereocenter count
-
Defined bond stereocenter count - bond stereocenter (or non-rotatable bond) was where two atoms could have different arrangement e.g. in cis- & trans- forms of butene around its double bond. Some compounds could have an undefined bond stereocenter (stereochemistry not specifically defined). Defined bond stereocenter count was the number of bond stereocenters where configurations were specifically defined.
-
Undefined bond stereocenter count - this was the undefined version of the bond stereocenter count
-
-
-
-
-
Convert data type for selected columns
-
-
# Convert data type - only for partition coefficients column (rest were okay)
-pc_cov = pc_cov.with_column((pl.col("Partition coefficients")).cast(pl.Float64, strict =False))
-pc_cov.head()
-
-
-
-
-shape: (5, 38)
-
-
-
-
-cid
-
-
-Compound name
-
-
-Synonyms
-
-
-Molecular weight
-
-
-Molecular formula
-
-
-Polar surface area
-
-
-Complexity
-
-
-Partition coefficients
-
-
-Heavy atom count
-
-
-Hydrogen bond donor count
-
-
-Hydrogen bond acceptor count
-
-
-Rotatable bond count
-
-
-inchi
-
-
-isosmiles
-
-
-canonicalsmiles
-
-
-inchikey
-
-
-iupacname
-
-
-Exact mass
-
-
-Monoisotopic mass
-
-
-Formal charge
-
-
-Covalently-bonded unit count
-
-
-Isotope atom count
-
-
-Total atom stereocenter count
-
-
-Defined atom stereocenter count
-
-
-Undefined atoms stereocenter count
-
-
-Total bond stereocenter count
-
-
-Defined bond stereocenter count
-
-
-Undefined bond stereocenter count
-
-
-pclidcnt
-
-
-gpidcnt
-
-
-MeSH headings
-
-
-annothits
-
-
-annothitcnt
-
-
-aids
-
-
-cidcdate
-
-
-sidsrcname
-
-
-depcatg
-
-
-annotation
-
-
-
-
-i64
-
-
-str
-
-
-str
-
-
-f64
-
-
-str
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-str
-
-
-f64
-
-
-f64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-str
-
-
-str
-
-
-i64
-
-
-str
-
-
-i64
-
-
-str
-
-
-str
-
-
-str
-
-
-
-
-
-
-5280453
-
-
-"Calcitriol"
-
-
-"calcitriol|322...
-
-
-416.6
-
-
-"C27H44O3"
-
-
-60.7
-
-
-688.0
-
-
-5.1
-
-
-30
-
-
-3
-
-
-3
-
-
-6
-
-
-"InChI=1S/C27H4...
-
-
-"C[C@H](CCCC(C)...
-
-
-"CC(CCCC(C)(C)O...
-
-
-"GMRQFYUYWCNGIN...
-
-
-"(1R,3S,5Z)-5-[...
-
-
-416.329
-
-
-416.329
-
-
-0
-
-
-1
-
-
-0
-
-
-6
-
-
-6
-
-
-0
-
-
-2
-
-
-2
-
-
-0
-
-
-22311
-
-
-46029
-
-
-"Calcitriol"
-
-
-"Biological Tes...
-
-
-12
-
-
-"485|631|731|78...
-
-
-20040916
-
-
-"A2B Chem|AA BL...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-9962735
-
-
-"Ubiquinol"
-
-
-"ubiquinol|992-...
-
-
-865.4
-
-
-"C59H92O4"
-
-
-58.9
-
-
-1600.0
-
-
-20.2
-
-
-63
-
-
-2
-
-
-4
-
-
-31
-
-
-"InChI=1S/C59H9...
-
-
-"CC1=C(C(=C(C(=...
-
-
-"CC1=C(C(=C(C(=...
-
-
-"QNTNKSLOFHEFPK...
-
-
-"2-[(2E,6E,10E,...
-
-
-864.7
-
-
-864.7
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-9
-
-
-9
-
-
-0
-
-
-2732
-
-
-21358
-
-
-"NULL"
-
-
-"Chemical and P...
-
-
-7
-
-
-"NULL"
-
-
-20061025
-
-
-"001Chemical|A2...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-5961
-
-
-"Glutamine"
-
-
-"L-glutamine|gl...
-
-
-146.14
-
-
-"C5H10N2O3"
-
-
-106.0
-
-
-146.0
-
-
--3.1
-
-
-10
-
-
-3
-
-
-4
-
-
-4
-
-
-"InChI=1S/C5H10...
-
-
-"C(CC(=O)N)[C@@...
-
-
-"C(CC(=O)N)C(C(...
-
-
-"ZDXPYRJPNDTMRX...
-
-
-"(2S)-2,5-diami...
-
-
-146.069
-
-
-146.069
-
-
-0
-
-
-1
-
-
-0
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-88218
-
-
-399
-
-
-"Glutamine"
-
-
-"Biological Tes...
-
-
-12
-
-
-"422|429|436|54...
-
-
-20040916
-
-
-"001Chemical|3B...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-2244
-
-
-"Aspirin"
-
-
-"aspirin|ACETYL...
-
-
-180.16
-
-
-"C9H8O4"
-
-
-63.6
-
-
-212.0
-
-
-1.2
-
-
-13
-
-
-1
-
-
-4
-
-
-3
-
-
-"InChI=1S/C9H8O...
-
-
-"CC(=O)OC1=CC=C...
-
-
-"CC(=O)OC1=CC=C...
-
-
-"BSYNRYMUTXBXSQ...
-
-
-"2-acetyloxyben...
-
-
-180.042
-
-
-180.042
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-127012
-
-
-364455
-
-
-"Aspirin"
-
-
-"Biological Tes...
-
-
-12
-
-
-"1|3|9|15|19|21...
-
-
-20040916
-
-
-"001Chemical|3B...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-457
-
-
-"1-Methylnicoti...
-
-
-"1-methylnicoti...
-
-
-137.16
-
-
-"C7H9N2O+"
-
-
-47.0
-
-
-136.0
-
-
--0.1
-
-
-10
-
-
-1
-
-
-1
-
-
-1
-
-
-"InChI=1S/C7H8N...
-
-
-"C[N+]1=CC=CC(=...
-
-
-"C[N+]1=CC=CC(=...
-
-
-"LDHMAVIPBRSVRG...
-
-
-"1-methylpyridi...
-
-
-137.071
-
-
-137.071
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-310
-
-
-674
-
-
-"NULL"
-
-
-"Biological Tes...
-
-
-8
-
-
-"61001|61002|14...
-
-
-20040916
-
-
-"001Chemical|3B...
-
-
-"Chemical Vendo...
-
-
-"COVID-19, COVI...
-
-
-
-
-
-
-
-
-
-
-
Select columns for data visualisations
-
The idea was really only keeping all the numerical columns for some data visualisations later. So I’ve dropped all the other columns in texts or of the string types.
-
-
# Drop unused columns in preparation for data visualisations
-pc_cov = pc_cov.drop([
-"cid",
-"Synonyms",
-"Molecular formula",
-"inchi",
-"isosmiles",
-"canonicalsmiles",
-"inchikey",
-"iupacname",
-"pclidcnt",
-"gpidcnt",
-"MeSH headings",
-"annothits",
-"annothitcnt",
-"aids",
-"cidcdate",
-"sidsrcname",
-"depcatg",
-"annotation"
-])
-
-pc_cov.head()
-
-
-
-
-shape: (5, 20)
-
-
-
-
-Compound name
-
-
-Molecular weight
-
-
-Polar surface area
-
-
-Complexity
-
-
-Partition coefficients
-
-
-Heavy atom count
-
-
-Hydrogen bond donor count
-
-
-Hydrogen bond acceptor count
-
-
-Rotatable bond count
-
-
-Exact mass
-
-
-Monoisotopic mass
-
-
-Formal charge
-
-
-Covalently-bonded unit count
-
-
-Isotope atom count
-
-
-Total atom stereocenter count
-
-
-Defined atom stereocenter count
-
-
-Undefined atoms stereocenter count
-
-
-Total bond stereocenter count
-
-
-Defined bond stereocenter count
-
-
-Undefined bond stereocenter count
-
-
-
-
-str
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-f64
-
-
-f64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-
-
-
-
-"Calcitriol"
-
-
-416.6
-
-
-60.7
-
-
-688.0
-
-
-5.1
-
-
-30
-
-
-3
-
-
-3
-
-
-6
-
-
-416.329
-
-
-416.329
-
-
-0
-
-
-1
-
-
-0
-
-
-6
-
-
-6
-
-
-0
-
-
-2
-
-
-2
-
-
-0
-
-
-
-
-"Ubiquinol"
-
-
-865.4
-
-
-58.9
-
-
-1600.0
-
-
-20.2
-
-
-63
-
-
-2
-
-
-4
-
-
-31
-
-
-864.7
-
-
-864.7
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-9
-
-
-9
-
-
-0
-
-
-
-
-"Glutamine"
-
-
-146.14
-
-
-106.0
-
-
-146.0
-
-
--3.1
-
-
-10
-
-
-3
-
-
-4
-
-
-4
-
-
-146.069
-
-
-146.069
-
-
-0
-
-
-1
-
-
-0
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-
-
-"Aspirin"
-
-
-180.16
-
-
-63.6
-
-
-212.0
-
-
-1.2
-
-
-13
-
-
-1
-
-
-4
-
-
-3
-
-
-180.042
-
-
-180.042
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-
-
-"1-Methylnicoti...
-
-
-137.16
-
-
-47.0
-
-
-136.0
-
-
--0.1
-
-
-10
-
-
-1
-
-
-1
-
-
-1
-
-
-137.071
-
-
-137.071
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-
-
-
-
-
-
-
-
-
Quick summary statistics of columns
-
-
# Overall descriptive statistics of kept columns
-pc_cov.describe()
-
-
-
-
-shape: (7, 21)
-
-
-
-
-describe
-
-
-Compound name
-
-
-Molecular weight
-
-
-Polar surface area
-
-
-Complexity
-
-
-Partition coefficients
-
-
-Heavy atom count
-
-
-Hydrogen bond donor count
-
-
-Hydrogen bond acceptor count
-
-
-Rotatable bond count
-
-
-Exact mass
-
-
-Monoisotopic mass
-
-
-Formal charge
-
-
-Covalently-bonded unit count
-
-
-Isotope atom count
-
-
-Total atom stereocenter count
-
-
-Defined atom stereocenter count
-
-
-Undefined atoms stereocenter count
-
-
-Total bond stereocenter count
-
-
-Defined bond stereocenter count
-
-
-Undefined bond stereocenter count
-
-
-
-
-str
-
-
-str
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-
-
-
-
-"count"
-
-
-"631"
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-631.0
-
-
-
-
-"null_count"
-
-
-"0"
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-173.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-
-
-"mean"
-
-
-null
-
-
-549.539675
-
-
-163.915368
-
-
-864.755626
-
-
-2.25917
-
-
-37.770206
-
-
-4.066561
-
-
-9.210777
-
-
-9.518225
-
-
-549.095022
-
-
-549.06013
-
-
--0.004754
-
-
-1.578447
-
-
-0.006339
-
-
-4.017433
-
-
-3.551506
-
-
-0.465927
-
-
-0.381933
-
-
-0.343899
-
-
-0.038035
-
-
-
-
-"std"
-
-
-null
-
-
-455.236826
-
-
-192.256415
-
-
-1000.220379
-
-
-3.926459
-
-
-31.821967
-
-
-6.348004
-
-
-8.694184
-
-
-15.393131
-
-
-455.064211
-
-
-454.958033
-
-
-0.358537
-
-
-1.610416
-
-
-0.079429
-
-
-6.128363
-
-
-5.787792
-
-
-2.364089
-
-
-1.181171
-
-
-1.107245
-
-
-0.363159
-
-
-
-
-"min"
-
-
-"(+)-Mefloquine...
-
-
-103.1
-
-
-0.0
-
-
-0.0
-
-
--24.0
-
-
-1.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-103.04
-
-
-103.04
-
-
--6.0
-
-
-1.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-
-
-"max"
-
-
-"sodium;8-amino...
-
-
-4114.0
-
-
-1650.0
-
-
-9590.0
-
-
-20.2
-
-
-291.0
-
-
-57.0
-
-
-65.0
-
-
-151.0
-
-
-4112.12
-
-
-4111.12
-
-
-2.0
-
-
-21.0
-
-
-1.0
-
-
-39.0
-
-
-39.0
-
-
-31.0
-
-
-11.0
-
-
-11.0
-
-
-7.0
-
-
-
-
-"median"
-
-
-null
-
-
-435.9
-
-
-110.0
-
-
-635.0
-
-
-2.5
-
-
-30.0
-
-
-3.0
-
-
-7.0
-
-
-6.0
-
-
-435.227
-
-
-435.227
-
-
-0.0
-
-
-1.0
-
-
-0.0
-
-
-1.0
-
-
-1.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-0.0
-
-
-
-
-
-
-
-
-
-
-
Conditional assignments in Polars
-
The longer I’ve used Polars, the more I like its coding styles of chaining a string of different code functions together to manipulate dataframes in one go. This usually might mean that we could avoid writing some repeated loop functions to achieve the same results. In the example below, I’d like to show how to chain “when-then-otherwise” expressions by using Polars.
-
-
-
Chaining when-then-otherwise expressions - creating groups in data
-
I had the idea of separating all data into 3 different ranges of partition coefficients, so that this could be shown visually in plots. One of the possible ways (other than writing a loop function), or really the long way, to do this might be like the code shown below:
A shorter and probably more elegant way was to use the “when-then-otherwise” expression in Polars for conditional assignments (the following code snippet was adapted with thanks to the author of Polars, Ritchie Vink and also the good old Stack Overflow):
-
-
pc_cov = pc_cov.with_column(
- pl.when((pl.col("Partition coefficients") <=-10))
- .then("Smaller than -10")
- .when((pl.col("Partition coefficients") >=-11) & (pl.col("Partition coefficients") <=5))
- .then("Between -11 and 5")
- .otherwise("Larger than 6")
- .alias("Part_coef_group")
-)
-
-pc_cov.head(10)
-
-# a new column would be added to the end of the dataframe
-# with a new column name, "Part_coef_group"
-# (scroll to the very right to see the added column)
-
-
-
-
-shape: (10, 21)
-
-
-
-
-Compound name
-
-
-Molecular weight
-
-
-Polar surface area
-
-
-Complexity
-
-
-Partition coefficients
-
-
-Heavy atom count
-
-
-Hydrogen bond donor count
-
-
-Hydrogen bond acceptor count
-
-
-Rotatable bond count
-
-
-Exact mass
-
-
-Monoisotopic mass
-
-
-Formal charge
-
-
-Covalently-bonded unit count
-
-
-Isotope atom count
-
-
-Total atom stereocenter count
-
-
-Defined atom stereocenter count
-
-
-Undefined atoms stereocenter count
-
-
-Total bond stereocenter count
-
-
-Defined bond stereocenter count
-
-
-Undefined bond stereocenter count
-
-
-Part_coef_group
-
-
-
-
-str
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-f64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-f64
-
-
-f64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-i64
-
-
-str
-
-
-
-
-
-
-"Calcitriol"
-
-
-416.6
-
-
-60.7
-
-
-688.0
-
-
-5.1
-
-
-30
-
-
-3
-
-
-3
-
-
-6
-
-
-416.329
-
-
-416.329
-
-
-0
-
-
-1
-
-
-0
-
-
-6
-
-
-6
-
-
-0
-
-
-2
-
-
-2
-
-
-0
-
-
-"Larger than 6"
-
-
-
-
-"Ubiquinol"
-
-
-865.4
-
-
-58.9
-
-
-1600.0
-
-
-20.2
-
-
-63
-
-
-2
-
-
-4
-
-
-31
-
-
-864.7
-
-
-864.7
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-9
-
-
-9
-
-
-0
-
-
-"Larger than 6"
-
-
-
-
-"Glutamine"
-
-
-146.14
-
-
-106.0
-
-
-146.0
-
-
--3.1
-
-
-10
-
-
-3
-
-
-4
-
-
-4
-
-
-146.069
-
-
-146.069
-
-
-0
-
-
-1
-
-
-0
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-"Between -11 an...
-
-
-
-
-"Aspirin"
-
-
-180.16
-
-
-63.6
-
-
-212.0
-
-
-1.2
-
-
-13
-
-
-1
-
-
-4
-
-
-3
-
-
-180.042
-
-
-180.042
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-"Between -11 an...
-
-
-
-
-"1-Methylnicoti...
-
-
-137.16
-
-
-47.0
-
-
-136.0
-
-
--0.1
-
-
-10
-
-
-1
-
-
-1
-
-
-1
-
-
-137.071
-
-
-137.071
-
-
-1
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-"Between -11 an...
-
-
-
-
-"Losartan"
-
-
-422.9
-
-
-92.5
-
-
-520.0
-
-
-4.3
-
-
-30
-
-
-2
-
-
-5
-
-
-8
-
-
-422.162
-
-
-422.162
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-"Between -11 an...
-
-
-
-
-"Vitamin E"
-
-
-430.7
-
-
-29.5
-
-
-503.0
-
-
-10.7
-
-
-31
-
-
-1
-
-
-2
-
-
-12
-
-
-430.381
-
-
-430.381
-
-
-0
-
-
-1
-
-
-0
-
-
-3
-
-
-3
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-"Larger than 6"
-
-
-
-
-"Nicotinamide"
-
-
-122.12
-
-
-56.0
-
-
-114.0
-
-
--0.4
-
-
-9
-
-
-1
-
-
-2
-
-
-1
-
-
-122.048
-
-
-122.048
-
-
-0
-
-
-1
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-"Between -11 an...
-
-
-
-
-"Adenosine"
-
-
-267.24
-
-
-140.0
-
-
-335.0
-
-
--1.1
-
-
-19
-
-
-4
-
-
-8
-
-
-2
-
-
-267.097
-
-
-267.097
-
-
-0
-
-
-1
-
-
-0
-
-
-4
-
-
-4
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-"Between -11 an...
-
-
-
-
-"Inosine"
-
-
-268.23
-
-
-129.0
-
-
-405.0
-
-
--1.3
-
-
-19
-
-
-4
-
-
-7
-
-
-2
-
-
-268.081
-
-
-268.081
-
-
-0
-
-
-1
-
-
-0
-
-
-4
-
-
-4
-
-
-0
-
-
-0
-
-
-0
-
-
-0
-
-
-"Between -11 an...
-
-
-
-
-
-
-
-
-
-
-
-
Import Plotly
-
Time for some data vizzes - importing Plotly first.
-
-
import plotly.express as px
-
-
-
-
-
Some examples of data visualisations
-
Below were some of the examples of building plots by using Plotly.
-
-
Partition coefficients vs. Molecular weights
-
-
fig = px.scatter(x = pc_cov["Partition coefficients"],
- y = pc_cov["Molecular weight"],
- hover_name = pc_cov["Compound name"],
- color = pc_cov["Part_coef_group"],
- width =800,
- height =400,
- title ="Partition coefficients vs. molecular weights for compounds used in COVID-19 clinical trials")
-
-fig.update_layout(
- title =dict(
- font =dict(
- size =15)),
- title_x =0.5,
- margin =dict(
- l =20, r =20, t =40, b =3),
- xaxis =dict(
- tickfont =dict(size =9),
- title ="Partition coefficients"
- ),
- yaxis =dict(
- tickfont =dict(size =9),
- title ="Molecular weights"
- ),
- legend =dict(
- font =dict(
- size =9)))
-
-fig.show()
-
-
-
-
-
-
-
-
-
Molecular weights vs. Complexity
-
-
fig = px.scatter(x = pc_cov["Molecular weight"],
- y = pc_cov["Complexity"],
- hover_name = pc_cov["Compound name"],
-#color = pc_cov["Part_coef_group"],
- width =800,
- height =400,
- title ="Molecular weights vs. complexity for compounds used in COVID-19 clinical trials")
-
-fig.update_layout(
- title =dict(
- font =dict(
- size =15)),
- title_x =0.5,
- margin =dict(
- l =20, r =20, t =40, b =3),
- xaxis =dict(
- tickfont =dict(size =9),
- title ="Molecular weights"
- ),
- yaxis =dict(
- tickfont =dict(size =9),
- title ="Complexity"
- ),
- legend =dict(
- font =dict(
- size =9)))
-
-fig.show()
-
-
-
-
-
-
-
-
-
-
Export prepared dataset
-
Two of the possible options to export the dataset for use in a Shiny app could be:
-
-
Convert Polars dataframe into a Pandas dataframe, so that it could be imported into the app for use (Polars not directly supported in Shiny for Python yet, but we could use its to_pandas() function to coerce an object e.g. a dataframe to be converted into a Pandas dataframe).
-
Another option was to save Polars dataframe as .csv file, then read in this file in the app.py script by using Pandas (which was the method I used for this particular app)
-
-
```{python}
-# --If preferring to use Pandas--
-# Convert Polars df into a Pandas df if needed
-df_name = df_name.to_pandas()
-
-# Convert the Pandas df into a csv file using Pandas
-df_name.to_csv("csv_file_name.csv", sep =",")
-
-# --If preferring to use Polars--
-# Simply write a Polars dataframe into a .csv file
-df_name.write_csv("csv_file_name.csv", separator =",")
-```
-
-
-
-
- ]]>
- Python
- Shiny
- Polars
- Plotly
- PubChem
- Cheminformatics
- https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_data_prep.html
- Sun, 07 May 2023 12:00:00 GMT
-
-
- Shiny app in R
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/12_Shiny_app_chembl/ShinyAppChembl.html
-
-
Back story
-
It has been a long while since I’ve used R in my projects. Last year during the time when I bravely embraced the role of rotating curator for @WeAreRLadies on Twitter around end of October, I did mention that I wanted to learn Shiny. I haven’t forgotten about this actually. So as promised, here it is, my first Shiny app in R, which is really a very simple interactive web app about small molecules from ChEMBL database. The URL to reach this app, ShinyAppChembl, is at: https://jhylin.shinyapps.io/ShinyAppChembl/. It shows a selected set of physicochemical properties for the curated small molecules in different max phases in boxplot formats. Note: it may take a few minutes to load the plot when first opening the app.
-
-
-
-
The process
-
Since I haven’t been using a lot of R lately, I just wanted to document how I approached this Shiny app framework, as part of my self-directed learning for R that started around mid-2022. The first place I went to was not Google’s Bard or OpenAI’s ChatGPT, as I was trying to preempt a scenario where if both of these options were temporarily down, what would I do to learn a new tool. So I visited the Shiny website first, and literally started from the “Get Started” section there, then tried to read through the lessons provided. I gathered a quick overview about the core components within a Shiny app, which were the user interface, server logic and the call to run or create app in the end, and thought to get started from there.
-
One of the most helpful online books called, “Mastering Shiny” had clarified a few coding issues for me. The reactivity section in the book was very useful as well to help with understanding the interactivity concept in the app. The best and also the hardest thing at this stage after reading some of the information was to actually start coding for the app in RStudio IDE, which I did soon after.
-
-
-
-
Trials-and-errors
-
Initially, I’ve noticed in the gallery section from the Shiny website that some of the basic Shiny apps had plots produced with R code using S3 method - the type with class ‘formula’, such as boxplot(formula e.g. y ~ group, data, and so on). So I started with this first and ended up with a draft version shown below:
-
-
-
-First draft app using S3 method in R code to plot boxplots - screenshot taken by author
-
-
-
-
I then tried the ggplot2 version, which I preferred to use. However, I kept on hitting a roadblock repeatedly (as shown in the image below):
-
-
-
-Second draft app using ggplot2 boxplot format - screenshot taken by author
-
-
-
I ended up working through this issue of not being able to display the boxplots properly over at least two days, where I tried to figure out how to change the code so that the boxplots would appear as the output in the app. I actually wrote a plot function code (as shown below) before working on the app.R file, in order to trial plotting the boxplots, making sure that the code worked before using it in the app.R file.
-
```{r}
-dfBoxplot <-function(var) {
- label <- rlang::englue("{{var}} vs. Max Phases of small molecules")
-
- chembl %>%
-select(`Max Phase`, {{ var }}) %>%
-ggplot(aes(x =`Max Phase`, y = {{ var }})) +
-geom_boxplot(aes(group =cut_width(`Max Phase`, 0.25),
-colour =`Max Phase`),
-outlier.alpha =0.2) +
-labs(title = label)
-}
-```
-
Once I made sure this code worked, I transplanted the code into the server section of the app.R file, however it wasn’t that simple obviously. Through the process of more trials-and-errors, I managed to figure out the code for the plot output in the final version, which was not the same as the function code above, but more like this.
-
```{r}
- output$BPlot <-renderPlot({
-
-ggplot(chembl, aes(`Max Phase`, .data[[input$variable]])) +
-geom_boxplot(aes(group =cut_width(`Max Phase`, 0.25),
-colour =`Max Phase`), outlier.alpha =0.2) +
-labs(title ="Distributions of physicochemical properties against max phases",
-caption ="(based on ChEMBL database version 31)") +
-theme_minimal()
-
- }, res =96) %>%bindCache(chembl$`Max Phase`, input$variable)
-```
-
I then read about the section on “Tidy evaluation” in the “Mastering Shiny” book, which had thoroughly described the problems I’ve encountered (and which I wished I had actually read this section before and not after hitting the roadblock…). So I’d highly recommend new users to read this section and also the rest of the book if Shiny’s also new to you.
-
-
-
-
Final app
-
The final app now looks like this:
-
-
-
-Screenshot taken by author
-
-
-
-
-
-
App deployment
-
After I got the app working, I looked into where I could deploy the app, since my main goal was to learn and share my work. At first, I went to the Shiny section on the Quarto website to see if it was possible to deploy the app in Quarto. However, after reading through several questions and answers in relation to Shiny apps and Quarto website, it was obvious that it was still not possible yet to deploy the app in an interactive way on Quarto websites (but it was mentioned in Posit community that this was being looked into, so I’m looking forward to the day when we can do exactly that in the future). This means that currently, there will only be an app image showing up in a Quarto document at most. I ended up choosing shinyapp.io to deploy my first Shiny app for now.
-
-
-
-
About the boxplots
-
Since the main goal of this post is more on the process of producing a simple Shiny app for a new comer, I won’t go into the fine details to describe how these boxplots differ between different max phases. Also as a side note, I’m aware that some experts in data visualisations might not really like boxplots in general, but for my case, I’ve got molecules in different max phases where a boxplot is presented for each max phase lining up next to each other. Therefore, in a way, some relative comparisons or differences could be drawn visually in the first glance, although other graph types such as density plots or heat maps might be better options.
-
I’ll focus on the “QED Weighted” variable here, as it’s a physicochemical property that has combined several molecular features together as a score (please refer to this post - section: “Some exploratory data analysis” for details about this QED weighted score). For all the boxplots shown when “QED Weighted” is selected from the drop-down box, max phase 4 molecules obviously have higher QED weighted scores in general than all of the other max phases. This is especially clear when comparing the medians between them, with max phase 4 small molecules having a median QED weighted score of more than 0.5, and the rest of the other max phases had 0.5 or below. The higher the QED weighted scores, the more druglike the molecules will be, and for max phase 4 molecules, they are mostly prescription medicines that have already reached approval and are already being widely prescribed. So this makes sense as this is being reflected in the boxplots for these ChEMBL small molecules.
-
-
-
-
Final words
-
Finally, I’m quite pleasantly surprised that there is also a Shiny in Python version, which has a Shinylive option to deploy Shiny app interactively in GitHub Gist and so on… I’ll most likely need to read further into this and make this as my next project. This is also a nice break from my recent machine learning projects, which I’ll try to return to once I’ve had enough fun with Shiny!
-
Thanks for reading.
-
-
-
-
- ]]>
- R
- Shiny
- ChEMBL database
- Cheminformatics
- https://jhylin.github.io/Data_in_life_blog/posts/12_Shiny_app_chembl/ShinyAppChembl.html
- Thu, 06 Apr 2023 12:00:00 GMT
-
-
-
- Pills dataset - Part 3
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_evcxr_polars_plotly_final.html
-
-
Background
-
The aim of this final part (part 3) for the pills dataset was really for me to start using Rust in a beginner-friendly way. Overall, this trilogy (parts 1 - 3) for the pills dataset formulated an overview of how to use Polars in Python (mainly), Pandas in Python (smaller section) and Polars in Rust (even little less as this was new to me) with Plotly. Over time, I’ve been finding myself learning more optimally by doing and applying, rather than just reading and thinking, so I’ve got myself started in this very new programming language, Rust, to get some familiarities. I anticipated that I would still work with Python and R mainly in the near future, so that I’m not diverting too much and would be at least proficient in at least one programming language.
-
My very initial idea was to integrate Rust-Polars, Plotly in Rust (Plotly.rs) and Jupyter-Evcxr together, and see if I could get a simple data visualisation out of a small dataset. Although the idea sounded simple enough, I was actually quite stuck at the step of importing one of the columns as x-axis variables in Rust-Polars to Plotly.rs. I figured it might possibly be due to my very lack-of-knowledge and lack-of-familiarities with Rust (I do need to continue reading the Rust programming language book), Polars (I’m better with Python-Polars actually), Plotly.rs and also Evcxr. Another possibility could be that Plotly.rs mainly had ndarray support, and Polars was not mentioned explicitly in Plotly.rs so my guess was that these two might not flow very well together. Also, Polars itself was constantly evolving and growing as well.
-
So I’ve decided to leave things as how it would be for now, before I delayed this post any further. If I happened to figure out how to do this in the future, then I’ll come back to update this last part of the project. While I was tackling this little issue mentioned above, somehow I’ve managed to deconstruct Polars dataframe in Rust in Evcxr. So I’ll show a little bit about it below. One slightly good news that came out from all of this, was that I’ve managed to import the other column as y-axis variables, which contained numbers, without problems. I’ve also figured out the Rust code to convert Series/ChunkedArray to vectors in Rust IDEs (e.g. VS Code, and quite a few others). So I did learn a few things while completing this post, and hoped I could expand further on this later.
-
Note: I’ve published all Rust code as print-only in Quarto markdown file, since it’s not possible to run them in RStudio IDE (Rust was not supported). So all Rust code were originally run on Jupyter Lab in MacOS, with code outputs being captured as screenshots, which were shown as photos in this post. Here’s the link to the .ipynb file in the GitHub repository for this portfolio website (or alternatively, you could access it from the GitHub icon link at the top of the web page), in case anyone wanted to see the full .ipynb version.
-
-
-
-
Import dependencies
-
These dependencies were known as crates in the world of Rust. I’d also like to think of them as libraries or packages we would install or import in Python and R. So this step was necessary before I even started anything decent in Rust. Similar things would also apply to Rust IDEs as well since I’ve played a little bit in VS Code previously.
-
```{rust}
-// Set up required dependencies
-:dep ndarray ="0.15.6"
-```
-
```{rust}
-:dep plotly = { version =">=0.8.0", features = ["plotly_ndarray"]}
-```
-
```{rust}
-// May take a few minutes to load polars crate (might depend on your machine specs)
-:dep polars = { version =">=0.26.0", features = ["lazy", "csv-file", "strings", "dtype-duration", "dtype-categorical", "concat_str", "rank", "lazy_regex", "ndarray"]}
-```
I’ve tested plotting in Plotly.rs after a few trials and errors at the beginning, but luckily I’ve spotted the ndarray support from the Plotly.rs book soon enough to figure out that I could convert the “count” column into a ndarray first, which was shown in the code below.
-
```{rust}
-// Switch Polars dataframe into 2D array
-// Ensure "ndarray" was added as one of the features for polars under dependencies
-
-/*Example from Polars documentation:
-let df = DataFrame::new(vec![a, b]).unwrap();
-let ndarray = df.to_ndarray::<Float64Type>().unwrap();
-println!("{:?}", ndarray);
-*/
-
-//Note: ndarray for numbers only, not strings, so only "count" column was converted
-let ndarray = df.to_ndarray::<Float64Type>().unwrap();
-println!("{:?}", ndarray);
-```
-
-
-
-Photo by author
-
-
-
-
-
-
Deconstructing Polars dataframe in Rust
-
Because “to_ndarray” was only for numerics and not strings, I ran into a problem trying to figure out how to best import this other “Colour” column into Plotly.rs. This led to my little convoluted journey to work with Polars dataframe in Rust, trying to see if I could convert the “Colour” column into a vector (which might not be the best way to do it, but as part of my Rust learning, I went for it anyway). I’ve subsequently tried plotting the “count” column in ndarray as a vector with success, based on the reference from Plotly.rs book that variables for x or y-axis could be placed into a vector by using a vector macro. Eventually, I didn’t quite achieve my goal but I’ve managed to break down or convert the Polars dataframe into different formats.
-
```{rust}
-// Select specific column or series by position
-let Colours = df[0].clone();
-
-//Alternative way to select specific column or series by name
-//let u =df.select_series(&["Colour"]);
-```
-
```{rust}
-Colours
-```
-
-
-
-Photo by author
-
-
-
There was a mention of storing series (column) in a vec (as series vector, not vector for strings) in Polars’ documentation, which I’ve tried to plot in Plotly.rs, but it unfortunately failed to work. One of my guesses could be due to the data type used for vector, as Rust was a very type-specific programming language, which also brought its well-known memory safety and other benefits in the long run. My immediate thought was that it probably needed to be a vector for strings, not series, which might make it work. Then I was searching on StackOverflow for similar questions and answers, then I found something related to what I wanted to do from Polars documentations as shown below.
-
```{rust}
-// Adapted from: https://docs.rs/polars/latest/polars/docs/eager/index.html#series
-// Extracting data:
-// To be able to extract data out of Series,
-// either by iterating over them or converting them to other datatypes like a Vec<T>,
-// we first need to downcast them to a ChunkedArray<T>.
-// This is needed because we don't know the data type that is held by the Series.
-
-/*use polars::prelude::*;
- use polars::df;
-
- fn extract_data() -> PolarsResult<()> {
- let df = df! [ "a" => [None, Some(1.0f32), Some(2.0)], "str" => ["foo", "bar", "ham"]]?;
-
-// first extract ChunkedArray to get the inner type.
-
- let ca = df.column("a")?.f32()?;
-
-// Then convert to vec
-
- let _to_vec: Vec<Option<f32>> = Vec::from(ca);
-
-// We can also do this with iterators
-
- let ca = df.column("str")?.utf8()?;
- let _to_vec: Vec<Option<&str>> = ca.into_iter().collect();
- let _to_vec_no_options: Vec<&str> = ca.into_no_null_iter().collect();
-
- Ok(())
-
-}*/
-```
-
Initially, I trialled the iterator function first.
-
```{rust}
-// Print out items in column by applying an iterator to it
-println!("{}", &Colours.iter().format("\n"));
-```
-
-
-
-Photo by author
-
-
-
Then, it took me quite a long time to just downcast Series into ChunkedArray, but somehow I’ve managed to figure out the code myself below. One of the likely reasons was due to my choice of using Evcxr, which required Rust code in slightly different formats than the ones in Rust IDEs (although almost the same).
-
```{rust}
-// Somehow worked out how to convert series to chunkedarray by accident!
-println!("{:?}", Colours.utf8().unwrap());
-```
-
-
-
-Photo by author
-
-
-
Then I moved onto trying to figure out how to convert or place a ChunkedArray into a vector, with the closest answer shown below. However, bear in mind that these Rust code were for Rust IDEs, and not for Evcxr, so this added slightly more complexities to what I was trying to do (perhaps I should just stick with Rust IDEs in the future…).
-
```{rust}
-//Adpated from StackOverflow - How to get a Vec from polars Series or ChunkedArray?
-//You can collect the values into a Vec.
-
-/*use polars::prelude::*;
-
-fn main() -> Result<()> { let s = Series::new("a", 0..10i32);
-
- let as_vec: Vec<Option<i32>>=s.i32()?.into_iter().collect();
-
-//if we are certain we don't have missing values
- let as_vec: Vec<i32> = s.i32()?.into_no_null_iter().collect();
- Ok(())
-
-}*/
-```
-
I also found another way to iterate the ChunkedArray.
I also found out, randomly, how to slice strings for Series in Polars. By changing the number of letters to slice through in Some(), the strings or words would vary length accordingly. Here, I’ve used “15” so it covered all the colours (note: the longest would be 12 characters for combination colours).
-
```{rust}
-// Another method to use if needing to slice strings
-let x =Colours.utf8().unwrap().str_slice(0, Some(15));
-x
-```
-
-
-
-Photo by author
-
-
-
Lastly, before I got too carried away, I just wanted to show the method from Polars documentations that this was the Polars’ way to select a specific column from Polars dataframe.
-
```{rust}
-let ca =df.clone().lazy().select([cols(["Colour"])]).collect()?;
-ca
-```
-
-
-
-Photo by author
-
-
-
-
-
-
Plotting Polars dataframe in Plotly.rs
-
For the x-axis, eventually, I reverted for manual input due to the issue mentioned in the background section. So the colours from the “Colour” column were stored in a vector set up manually, rather than coming directly from the dataframe. While searching for answers, I’ve also learnt several other tricks, although not really solving the problem, they might still be useful in the future. For the y-axis, the ndarray for the “count” column was converted into a vector first before being fed into the trace (graph module), and thankfully the plot worked nicely.
-
```{rust}
-// MANUAL method:
-// Use vec! macro to create new vectors to hold x variables (words as strings)
-// Manually input the colour names (as ndarray is only for numbers)
-let x = vec!["RED", "ORANGE;BROWN", "YELLOW;WHITE", "ORANGE", "WHITE", "BLUE"];
-
-// Plot using ndarray, which is supported by Plotly.rs
-// Polars likely not supported yet
-// Convert ndarray (holding counts as y variables) into vector
-let y =ndarray.column(1).to_vec();
-
-// Use trace as a graph module,
-// choose type of plots needed for x & y variables called
-// Graph options e.g. Scatter, Line or Bar
-let trace = Scatter::new(x, y);
-
-// Set plot variable as mutable and initiate a plot
-let mut plot = Plot::new();
-// Add trace (graph) into the plot variable
-plot.add_trace(trace);
-
-// Specify the specs for plot
-let layout = Layout::new()
-// Choose height of graph
-.height(500)
-// Name x-axis
-.x_axis(Axis::new().title(Title::new("Colours")))
-// Name y-axis
-.y_axis(Axis::new().title(Title::new("Count")))
-// Add title of graph
-.title(Title::new("Frequency of colours in acetaminophen (paracetamol) oral dosage forms"));
-
-// Set the layout of the plot
-plot.set_layout(layout);
-
-// Display the plot in Jupyter Lab format
-// For Jupyter Notebook, use:plot.notebook_display();
-plot.lab_display();
-format!("EVCXR_BEGIN_CONTENT application/vnd.plotly.v1+json\n{}\nEVCXR_END_CONTENT", plot.to_json())
-```
-
-
-
-Photo by author
-
-
-
-
-
-
Conclusion
-
This last part was the hardest for me to execute out of all 3 parts (it likely took me a good whole week to figure out deconstructing Polars dataframe and trying to work with vectors), as Rust was completely new to me. At one point I thought about jumping back to Python, but I persisted and although I didn’t quite solve the string importation issue, I was somehow happy that I was at least able to see how this programming language could be applied in Polars dataframe library. I also got a taste of using Rust in data visualisations. All I wanted to show was that there were a variety of data tools to use, and knowing your tools of trade would be the most critical when working on different data projects as certain tools would only work the best for certain tasks and scenarios. This warm-up lesson in Rust was quite interesting and I might continue either in VS Code or Evcxr depending on my next topic of interest.
Plotly.rs GitHub repository: https://github.com/igiagkiozis/plotly (link to the Plotly.rs book can be found in “Converting columns into ndarrays” section)
-
-
-
-
- ]]>
- Data analytics projects
- Pills dataset series
- Rust
- Polars
- Plotly
- Evcxr
- Jupyter
- https://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_evcxr_polars_plotly_final.html
- Mon, 13 Feb 2023 11:00:00 GMT
-
-
-
- Pills dataset - Part 2
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_polars_pills_df.html
-
-
Quick overview
-
Part 2 of this project aimed to look at the pills data up close, particularly into the types of dosage forms, colours, shapes and inactive excipients used in oral medications. Plotly was used as the main data visualisation library, which was followed by some text cleaning for a particularly busy column in the dataset. This was then completed with a section in the end to generate a small dataframe, preparing for a simple data visualisation in Rust-Evcxr for the final part of this project (part 3).
One way to avoid switching Polars dataframe to a Pandas one, which could be one of the options to plot data from Polars dataframes in Plotly, was to call the x-axis and y-axis data directly from the dataframe as shown in the code below.
-
-
# scatter plot for colours, dosage forms & drug strengths
-fig = px.scatter(x = df_viz["Colour"],
- y = df_viz["Dosage_form"],
- color = df_viz["Colour"],
- hover_name = df_viz["Drug_strength"],
- width =900,
- height =400,
- title ="Oral dosage forms and colours of pills")
-
-# Update layout of the plot
-fig.update_layout(
-# Change title font size
- title =dict(
- font =dict(
- size =15)),
-# Centre the title
- title_x =0.5,
-# Edit margins
- margin =dict(
- l =20, r =20, t =40, b =3),
-# Change x-axis
- xaxis =dict(
- tickfont =dict(size =9),
- title ="Colours"
- ),
-# Change y-axis
- yaxis =dict(
- tickfont =dict(size =9),
- title ="Dosage forms"
- ),
-# Edit lengend font size
- legend =dict(
- font =dict(
- size =9)))
-
-fig.show()
-
-
-
-
-
-
-
White was the most common colour, especially after zooming in the plot. Capsule was very commonly used as the oral dosage form of choice in this dataset.
-
-
-
-
Visualising shapes & colours in pills
-
-
fig = px.scatter(x = df_viz["Colour"],
- y = df_viz["Shape"],
- color = df_viz["Colour"],
- hover_name = df_viz["Drug_strength"],
- width =900,
- height =400,
- title ="Shapes and colours of pills")
-
-# Update layout of the plot
-fig.update_layout(
-# Change title font size
- title =dict(
- font =dict(
- size =15)),
-# Centre the title
- title_x =0.5,
-# Edit margins
- margin =dict(
- l =20, r =20, t =40, b =3),
-# Change x-axis
- xaxis =dict(
- tickfont =dict(size =9),
- title ="Colours"
- ),
-# Change y-axis
- yaxis =dict(
- tickfont =dict(size =9),
- title ="Shapes"
- ),
-# Edit lengend font size
- legend =dict(
- font =dict(
- size =9)))
-
-fig.show()
-
-
-
-
-
-
-
Capsule was again the most common oral dosage shape used for pills in the dataset. Common colours included red, brown, blue, purple, pink, orange, green, white and yellow. Combination colours followed these common ones, which had a mixture of a variety of colours used simultaneously, likely to avoid confusions and errors in dispensings or administrations.
-
-
-
-
Visualising inactive excipients in pills
-
The messiest part of the data actually lied in the column of “Inactive_excipients”, with numerous different punctuations used inconsistently, such as forward slashes, commas and semi-colons. There were vast quantities of different inactive components used for oral dosage forms. Because of this, I had to spend a bit more time cleaning up the texts in order to find out what were the commonly used inactive ingredients in the end.
-
-
# Formulated a separate dataframe with just "Inactive_excipients"
-df_ie = df_new.select([pl.col("Inactive_excipients")])
-df_ie
-
-
-
-
-shape: (83925, 1)
-
-
-
-
-Inactive_excipients
-
-
-
-
-str
-
-
-
-
-
-
-"SILICON DIOXID...
-
-
-
-
-"SILICON DIOXID...
-
-
-
-
-"ANHYDROUS LACT...
-
-
-
-
-null
-
-
-
-
-"SORBITOL;ASPAR...
-
-
-
-
-"ANHYDROUS DIBA...
-
-
-
-
-"LACTOSE MONOHY...
-
-
-
-
-"FD&C BLUE NO. ...
-
-
-
-
-"silicon dioxid...
-
-
-
-
-"CROSPOVIDONE;M...
-
-
-
-
-"STARCH, CORN;C...
-
-
-
-
-"COPOVIDONE K25...
-
-
-
-
-...
-
-
-
-
-"SILICON DIOXID...
-
-
-
-
-"BUTYLATED HYDR...
-
-
-
-
-"MAGNESIUM CARB...
-
-
-
-
-"ACESULFAME POT...
-
-
-
-
-"CROSCARMELLOSE...
-
-
-
-
-"FD&C BLUE NO. ...
-
-
-
-
-"STARCH, CORN;H...
-
-
-
-
-"CARNAUBA WAX;F...
-
-
-
-
-"CITRIC ACID MO...
-
-
-
-
-"STARCH, CORN;D...
-
-
-
-
-"Cellulose, mic...
-
-
-
-
-"ANHYDROUS DIBA...
-
-
-
-
-
-
-
-
-
Text cleaning for inactive excipients column
-
To prepare this column for data visualisations, I used Polars’ string expressions (or more commonly known as regex - regular expressions) to try and tidy up the raw texts. When I did the text cleaning in Jupyter Lab initially, the line of code for .str.strip(” ,“) worked, but when I converted the .ipynb file into a .qmd (Quarto markdown) one, and used the same line, it failed to work due to the extra space in front of the comma. However, I got around the error by splitting it into two separate units as space and comma, and it worked without problem. One possible reason would be due to the reticulate package needed to run Python in RStudio IDE, and how Polars dataframe library was relatively newer than Pandas dataframe library, which meant certain features in Polars might not have been taken on board in the reticulate package (only my guess).
-
-
# Clean string texts
-# Convert uppercase letters into lowercase ones in the excipients column
-df_de = (df_ie.with_column(pl.col("Inactive_excipients").str.to_lowercase(
-# replace old punctuations (1st position) with new one (2nd position)
- ).str.replace_all(
-";", ", "
- ).str.replace_all(
-" /", ", "
- ).str.replace_all(
-"/", ", "
-# Remove extra space & comma by stripping
-# In Jupyter notebook/lab - can combine space & comma: .str.strip(" ,")
-# For RStudio IDE - separate into two for this to work
- ).str.strip(
-" "
- ).str.strip(
-","
-# Split the texts by the specified punctuation e.g. comma with space
- ).str.split(
- by =", "
-# Create a new column with a new name
- ).alias(
-"Inactive"
- )
-# Explode the splitted texts into separate rows within the new column
-).explode(
-"Inactive"
-)
-)
-
-df_de
-
-
-
-
-shape: (840029, 2)
-
-
-
-
-Inactive_excipients
-
-
-Inactive
-
-
-
-
-str
-
-
-str
-
-
-
-
-
-
-"SILICON DIOXID...
-
-
-"silicon dioxid...
-
-
-
-
-"SILICON DIOXID...
-
-
-"edetate disodi...
-
-
-
-
-"SILICON DIOXID...
-
-
-"lactose monohy...
-
-
-
-
-"SILICON DIOXID...
-
-
-"magnesium stea...
-
-
-
-
-"SILICON DIOXID...
-
-
-"cellulose"
-
-
-
-
-"SILICON DIOXID...
-
-
-"microcrystalli...
-
-
-
-
-"SILICON DIOXID...
-
-
-"starch"
-
-
-
-
-"SILICON DIOXID...
-
-
-"corn"
-
-
-
-
-"SILICON DIOXID...
-
-
-"sodium lauryl ...
-
-
-
-
-"SILICON DIOXID...
-
-
-"fd&c blue no. ...
-
-
-
-
-"SILICON DIOXID...
-
-
-"fd&c red no. 4...
-
-
-
-
-"SILICON DIOXID...
-
-
-"gelatin"
-
-
-
-
-...
-
-
-...
-
-
-
-
-"Cellulose, mic...
-
-
-"shellac"
-
-
-
-
-"Cellulose, mic...
-
-
-"propylene glyc...
-
-
-
-
-"Cellulose, mic...
-
-
-"ammonia"
-
-
-
-
-"Cellulose, mic...
-
-
-"fd&c blue no. ...
-
-
-
-
-"ANHYDROUS DIBA...
-
-
-"anhydrous diba...
-
-
-
-
-"ANHYDROUS DIBA...
-
-
-"ferric oxide r...
-
-
-
-
-"ANHYDROUS DIBA...
-
-
-"hypromelloses"
-
-
-
-
-"ANHYDROUS DIBA...
-
-
-"polyethylene g...
-
-
-
-
-"ANHYDROUS DIBA...
-
-
-"magnesium stea...
-
-
-
-
-"ANHYDROUS DIBA...
-
-
-"titanium dioxi...
-
-
-
-
-"ANHYDROUS DIBA...
-
-
-"talc"
-
-
-
-
-"ANHYDROUS DIBA...
-
-
-"ferric oxide y...
-
-
-
-
-
-
-
-
-
# Quick look at the dataframe to see before and after text cleaning
-print(df_de.glimpse())
-
-
Rows: 840029
-Columns: 2
-$ Inactive_excipients <Utf8> SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;
-$ Inactive <Utf8> silicon dioxide, edetate disodium, lactose monohydrate, magnesium stearate, cellulose, microcrystalline, starch, corn, sodium lauryl sulfate, fd&c blue no. 1
-
-
-
-
As shown above, the “Inactive_excipients” column was the original column for excipients, where the second column named, “Inactive” was the new column shown after the punctuation tidy-ups, string strip and row text explosion. The excipients were broken down into individual terms, rather than in massively long strings which might not make sense to some readers.
-
-
# Re-organise the dataframe to choose the cleaned "Inactive" column
-df_final = df_de.select(["Inactive"])
-df_final
-
-
-
-
-shape: (840029, 1)
-
-
-
-
-Inactive
-
-
-
-
-str
-
-
-
-
-
-
-"silicon dioxid...
-
-
-
-
-"edetate disodi...
-
-
-
-
-"lactose monohy...
-
-
-
-
-"magnesium stea...
-
-
-
-
-"cellulose"
-
-
-
-
-"microcrystalli...
-
-
-
-
-"starch"
-
-
-
-
-"corn"
-
-
-
-
-"sodium lauryl ...
-
-
-
-
-"fd&c blue no. ...
-
-
-
-
-"fd&c red no. 4...
-
-
-
-
-"gelatin"
-
-
-
-
-...
-
-
-
-
-"shellac"
-
-
-
-
-"propylene glyc...
-
-
-
-
-"ammonia"
-
-
-
-
-"fd&c blue no. ...
-
-
-
-
-"anhydrous diba...
-
-
-
-
-"ferric oxide r...
-
-
-
-
-"hypromelloses"
-
-
-
-
-"polyethylene g...
-
-
-
-
-"magnesium stea...
-
-
-
-
-"titanium dioxi...
-
-
-
-
-"talc"
-
-
-
-
-"ferric oxide y...
-
-
-
-
-
-
-
-
-
# Remove all cells with null values
-df_final = df_final.drop_nulls()
-
-
-
# Group the data by different inactive excipients with counts shown
-df_final = df_final.groupby("Inactive").agg(pl.count())
-df_final.head()
-
-
-
-
-shape: (5, 2)
-
-
-
-
-Inactive
-
-
-count
-
-
-
-
-str
-
-
-u32
-
-
-
-
-
-
-"low-substitute...
-
-
-4
-
-
-
-
-"sodium starch ...
-
-
-118
-
-
-
-
-" glyceryl dibe...
-
-
-3
-
-
-
-
-"aluminum chlor...
-
-
-27
-
-
-
-
-"mentha piperit...
-
-
-7
-
-
-
-
-
-
-
-
-
-
Inactive excipient counts
-
-
# Count each excipient and cast the whole column into integers
-df_final = df_final.with_column((pl.col("count")).cast(pl.Int64, strict =False))
-df_final
-
-
-
-
-shape: (1674, 2)
-
-
-
-
-Inactive
-
-
-count
-
-
-
-
-str
-
-
-i64
-
-
-
-
-
-
-"low-substitute...
-
-
-4
-
-
-
-
-"sodium starch ...
-
-
-118
-
-
-
-
-" glyceryl dibe...
-
-
-3
-
-
-
-
-"aluminum chlor...
-
-
-27
-
-
-
-
-"mentha piperit...
-
-
-7
-
-
-
-
-"epimedium gran...
-
-
-1
-
-
-
-
-" ethyl acetate...
-
-
-2
-
-
-
-
-"rutin"
-
-
-1
-
-
-
-
-"methacrylic ac...
-
-
-2106
-
-
-
-
-" calcium phosp...
-
-
-12
-
-
-
-
-"carbomer homop...
-
-
-28
-
-
-
-
-" tocopherol"
-
-
-2
-
-
-
-
-...
-
-
-...
-
-
-
-
-"methylcellulos...
-
-
-62
-
-
-
-
-"carbomer homop...
-
-
-27
-
-
-
-
-" red ferric ox...
-
-
-3
-
-
-
-
-"anhydrous lact...
-
-
-4
-
-
-
-
-"sorbic acid"
-
-
-195
-
-
-
-
-"ilex pedunculo...
-
-
-2
-
-
-
-
-" aminobenzoic ...
-
-
-1
-
-
-
-
-"polyvinyl alco...
-
-
-55
-
-
-
-
-"3-hexenyl acet...
-
-
-4
-
-
-
-
-"methacrylic ac...
-
-
-2
-
-
-
-
-"dihydroxyalumi...
-
-
-2
-
-
-
-
-"hydroxypropyl ...
-
-
-46
-
-
-
-
-
-
-
-
-
-
Overview of inactive excipients used in oral dosage forms
-
-
fig = px.scatter(x = df_final["Inactive"],
- y = df_final["count"],
- hover_name = df_final["Inactive"],
- title ="Inactive excipients and their respective counts in pills")
-
-fig.update_layout(
- title =dict(
- font =dict(
- size =15)),
- title_x =0.5,
- margin =dict(
- l =20, r =20, t =40, b =10),
- xaxis =dict(
- tickfont =dict(size =9),
- title ="Inactive excipients"
- ),
- yaxis =dict(
- tickfont =dict(size =9),
- title ="Counts"
- ),
- legend =dict(
- font =dict(
- size =9)))
-
-
-fig.show()
-
-
-
-
-
-
-
-
Frequently used inactive excipients
-
-
# Re-order the excipients with counts in descending order
-# Filter out only the ones with counts over 10,000
-df_ex = df_final.sort("count", reverse =True).filter((pl.col("count")) >=10000)
-df_ex.head()
-
-
-
-
-shape: (5, 2)
-
-
-
-
-Inactive
-
-
-count
-
-
-
-
-str
-
-
-i64
-
-
-
-
-
-
-"magnesium stea...
-
-
-58908
-
-
-
-
-"titanium dioxi...
-
-
-43241
-
-
-
-
-"unspecified"
-
-
-35744
-
-
-
-
-"silicon dioxid...
-
-
-34037
-
-
-
-
-"starch"
-
-
-32501
-
-
-
-
-
-
-
-
-
fig = px.bar(x = df_ex["Inactive"],
- y = df_ex["count"],
- color = df_ex["Inactive"],
- title ="Commonly used inactive excipients in pills")
-
-fig.update_layout(
- title =dict(
- font =dict(
- size =15)),
- title_x =0.5,
- margin =dict(
- l =10, r =10, t =40, b =5),
- xaxis =dict(
- tickfont =dict(size =9),
- title ="Inactive excipients"
- ),
- yaxis =dict(
- tickfont =dict(size =9),
- title ="Counts"
- ),
- legend =dict(
- font =dict(
- size =9)))
-
-fig.show()
-
-
-
-
-
-
-
The text cleaning might not be perfect at this stage, but I think I’ve managed to get most of the core texts cleaned into a more sensible and readable formats. From what I’ve worked out here, the most frequently used inactive ingredient was magnesium stearate, which was followed by titanium dioxide, and then interestingly “unspecified”, which was exactly how it was documented in the original pillbox dataset at the beginning. I didn’t go further digging into what this “unspecified” inactive excipients might be, as in whether it meant it in a singular or plural forms. So this still remained a mystery at this stage, but if all these oral medications were FDA-approved, we would’ve hoped each and everyone of these pills would be verified in safety, quality and effectiveness before they entered into the market for wide prescriptions. In the worst case, each therapeutic drug should also have post-marketing surveillance, for long-term safety monitoring.
-
-
-
-
-
Create a small dataframe for data visualisation in Rust-Evcxr
-
All acetaminophens were filtered out in the “Drug_strength” column and all duplicates were removed in the dataset.
I’ve opted for finding out the different types of colours with their respective counts in oral acetaminophen, or also known as paracetamol in some other countries.
fig = px.scatter(x = df_ac["Colour"],
- y = df_ac["count"],
- size = df_ac["count"],
- color = df_ac["Colour"],
- title ="Frequency of colours in acetaminophen (paracetamol) oral dosage forms"
- )
-
-fig.update_layout(
- xaxis =dict(
- title ="Colours"
- ),
- yaxis =dict(
- title ="Counts"
- )
-)
-
-fig.show()
-
-
-
-
-
-
-
I’ve decided to keep the dataframe very simple for part 3 as my original intention was to trial plotting a graph in Evcxr only (nothing fancy at this stage), and also to gain some familiarities with Rust as another new programming language for me. Readers might notice that I’ve opted for a scatter plot in Plotly (in Python3 kernel) here for this last dataframe, and when we finally got to part 3 (hopefully coming soon as I needed to figure how to publish Rust code in Quarto…), I might very likely revert this to a bar graph (in Rust kernel), due to some technical issues (slow dependency loading, and somehow with Plotly.rs in Evcxr, the scatter graph looked more like scatter line graph instead… more stories to follow) and being a new Rust-Evcxr user. At the very least, I’ve kind of tried something I’ve planned for, although not looking very elegant yet, with rooms for improvements in the future.
-
-
-
-
- ]]>
- Data analytics projects
- Pills dataset series
- Polars
- Python
- Plotly
- Jupyter
- https://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_polars_pills_df.html
- Mon, 30 Jan 2023 11:00:00 GMT
-
-
-
- Pills dataset - Part 1
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_polars_pills_ws.html
-
-
Introduction
-
As mentioned in my last project, I’ve tried using Evcxr, which provided a way to use Rust interactively in a Jupyter environment. The name, “Evcxr”, was quite hard to remember at first. It was pronounced as “e-vic-ser” according to the author, which I’ve randomly come across in an online tech interview when I was looking into it. I’ve also sort of worked out a way to memorise its spelling by taking specific letters out of “evaluation context for rust” (which was what it was called in its GitHub repository).
-
For users of Jupyter Notebook/Lab and Python, they might be quite used to the working speed of the cell outputs. However, one thing I’ve noticed when I was using Evcxr or Rust kernel in Jupyter Lab was that the speed of cell outputs was noticeably slower (especially at the beginning while loading all the dependencies required). The speed improved when loading external crates and modules, and generally it was faster afterward.
-
Due to this reason (note: I did not look into any other optimising strategies for this and this could be restricted to my computer hardware specs, so this might differ for other users), I think Evcxr was not ideal for a very large and complex data science project yet (however if its ecosystem kept developing, it might be improved in the future). One thing of note was that when I was combing through issues in Evcxr’s GitHub repository, someone mentioned the slow compile time of the Rust compiler, which would have likely caused the snail speed, but knowing that the actual program running speed was blazingly fast, some sacrifice at the beginning made sense to me. Overall, Rust was really a systems programming language with memory efficiency (with no garbage collector), type safety and concurrency as some of its notable advantages.
-
Because of the dependency loading issue in the Jupyter environment, and also knowing there was already a dataframe library built from Rust, I’ve opted to use Polars-Python again for the data wrangling part of this project. This was also accompanied by the good old Pandas library as well (under the section of “Transform web-scraped data into dataframe” if anyone wants to jump to that part to see the code). I then went on to trial using Rust via Evcxr for data visualisation based on a small dataframe by using Plotly.rs. This project would be separated into 3 parts:
-
-
Part 1: Initial pillbox dataset loading and web-scraping
-
Part 2: Data wrangling and mining for data visualisations
-
Part 3: Using Rust for data visualisation
-
-
The main reason I wanted to try Evcxr was that I could see the potential of using Rust interactively to showcase the results in a relatively fast and efficient manner. This meant specific data exploratory results could reach wider audience, leading to more impacts in different fields, in a very broad term. Oppositely, for more specific users such as scientists or engineers, this meant experiments could be carried out in a safe and efficient manner, with test results readily available for future work planning.
-
-
-
-
Download dataset
-
This time the dataset was spotted from Data Is Plural, specifically the 2022.11.30 edition. The section I was interested in was the first paragraph at the top, about “Pills”. By going into one of the links provided in the paragraph, this brought me to the Pillbox dataset from the US National Library of Medicine (NLM). The .csv file was downloaded via the “Export” button at the top right of the webpage.
-
This pillbox dataset was actually retired since 28th January 2021, but was still available for educational or research purposes only. Therefore, it was not recommended for pill identifications as the dataset was not up-to-date. Alternative resources such as DailyMed would be more appropriate for readers in the US (as one of the examples). For readers in other countries, local health professionals and resources would be recommended for up-to-date information.
-
-
-
-
Importing library & dataset
-
-
# Install/upgrade polars if needed (uncomment the line below)
-#pip install --upgrade polars
-
-
-
import polars as pl
-
-
-
# Check version of polars (uncomment line below)
-#pl.show_versions()
When importing pillbox.csv file initially, an error message actually came up that showed, “…Could not parse ‘10.16’ as dtype Int64 at column 7…”. One way to get around this was to add “ignore_errors” to bypass this error first in order to load the dataset first. This error could be fixed when checking and converting data types for columns.
-
-
-
-
Initial data wrangling
-
The Pillbox dataset link from NLM provided a list of column information for users. To quickly see what were the columns in the dataset, we could use “df.glimpse()” to read column names, data types and the first 10 items in each column.
A relatively simple dataset would be extracted first for these pills data since I was an inexperienced user of Rust. Therefore, I’ve selected only certain columns for this purpose.
-
-
df_med = df.select([# shapes of medicines
-"splshape_text",
-# colours of medicines
-"splcolor_text",
-# strengths of medicines
-"spl_strength",
-# inactive ingredients/excipients in medicines
-"spl_inactive_ing",
-# dosage forms of medicines e.g. capsules or tablets etc.
-"dosage_form"]
- )
-df_med
This was not planned initially but this might make my life a lot easier if I could scrape the dosage form table found through the Pillbox link, since the dosage form column was full of C-letter code. These dosage form code were hard to understand, so once I’ve got the code along with corresponding dosage forms in texts, the web-scraped information would be converted into a dataframe for further data manipulations.
-
-
# Uncomment lines below to install libraries needed for web-scraping
-#!pip install requests
-#!pip install beautifulsoup4
-
-
-
Import libraries
-
-
import requests
-from bs4 import BeautifulSoup
-import pandas as pd
-
-
I’ve opted for using Beautiful Soup as the web-scraping library in Python, along with the requests library to be able to make a URL request call to retrieve web information. There were of course many other tools available as well. A caveat to be taken into consideration was that when web-scraping, it was always recommended to check whether the information being scraped were under a specific copyright license and so on. In this case, I’ve checked that the dosage form table link - https://www.fda.gov/industry/structured-product-labeling-resources/dosage-forms was from US FDA and it was stated that the information (both texts and graphs) were not copyrighted (unless otherwise stated, for this particular web page, there was nothing stated along those lines), but a link to this webpage should be provided so that readers could access most current information in the future.
-
-
-
Send web requests
-
-
# Specify URL address with information intended for web-scraping
-url ="https://www.fda.gov/industry/structured-product-labeling-resources/dosage-forms"
-# Request the web information via requests library & save under a data object
-data = requests.get(url)
-
-
-
-
Parse web content
-
-
# Parse the web content from the URL link by using Beautiful Soup
-soup = BeautifulSoup(data.content, "html.parser")
-
-
-
-
Print web content
-
-
# Print out the scraped web information
-print(soup.prettify())
## Get a list of moderate cyp3a4 inhibitors
+# skipping grapefruit juice as it's not quite an approved drug...
+# note: amlodipine inhibits cyp3a5
+df_3a4_mod_inh = chembl_drugs(
+"AMLODIPINE", "APREPITANT", "CIPROFLOXACIN", "CRIZOTINIB", "DILTIAZEM", "ERYTHROMYCIN", "FLUCONAZOLE", "IMATINIB", "LETERMOVIR", "NETUPITANT", "VERAPAMIL", #file_name="mod_3a4_inh"
+ )
+df_3a4_mod_inh.head()
+
+
+
+
+
+
+
+
+
chembl_id
+
pref_name
+
max_phase
+
canonical_smiles
+
+
+
+
+
0
+
CHEMBL1491
+
AMLODIPINE
+
4
+
CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1c1ccccc1Cl
+
+
+
1
+
CHEMBL1471
+
APREPITANT
+
4
+
C[C@@H](O[C@H]1OCCN(Cc2n[nH]c(=O)[nH]2)[C@H]1c...
+
+
+
2
+
CHEMBL8
+
CIPROFLOXACIN
+
4
+
O=C(O)c1cn(C2CC2)c2cc(N3CCNCC3)c(F)cc2c1=O
+
+
+
3
+
CHEMBL601719
+
CRIZOTINIB
+
4
+
C[C@@H](Oc1cc(-c2cnn(C3CCNCC3)c2)cnc1N)c1c(Cl)...
+
+
+
4
+
CHEMBL23
+
DILTIAZEM
+
4
+
COc1ccc([C@@H]2Sc3ccccc3N(CCN(C)C)C(=O)[C@@H]2...
+
+
+
+
+
+
+
+
# Get a list of strong cyp2d6 inhibitors
+df_2d6_strong_inh = chembl_drugs(
+"BUPROPION", "FLUOXETINE", "PAROXETINE", "QUINIDINE",
+#file_name="strong_2d6_inh"
+ )
+df_2d6_strong_inh
+
+
+
+
+
+
+
+
+
chembl_id
+
pref_name
+
max_phase
+
canonical_smiles
+
+
+
+
+
0
+
CHEMBL894
+
BUPROPION
+
4.0
+
CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1
+
+
+
1
+
CHEMBL41
+
FLUOXETINE
+
4.0
+
CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1
+
+
+
2
+
CHEMBL490
+
PAROXETINE
+
4.0
+
Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1
+
+
+
3
+
CHEMBL21578
+
QUINIDINE
+
NaN
+
C=C[C@H]1CN2CCC1C[C@@H]2[C@@H](O)c1ccnc2ccc(OC...
+
+
+
4
+
CHEMBL1294
+
QUINIDINE
+
4.0
+
C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc...
+
+
+
+
+
+
+
+
# Get a list of moderate cyp2d6 inhibitors
+df_2d6_mod_inh = chembl_drugs(
+"ABIRATERONE", "CINACALCET", "CLOBAZAM", "DOXEPIN", "DULOXETINE", "HALOFANTRINE", "LORCASERIN", "MOCLOBEMIDE", "ROLAPITANT", "TERBINAFINE",
+#file_name="mod_2d6_inh"
+ )
+df_2d6_mod_inh.head()
+
+
+
+
+
+
+
+
+
chembl_id
+
pref_name
+
max_phase
+
canonical_smiles
+
+
+
+
+
0
+
CHEMBL254328
+
ABIRATERONE
+
4
+
C[C@]12CC[C@H](O)CC1=CC[C@@H]1[C@@H]2CC[C@]2(C...
+
+
+
1
+
CHEMBL1201284
+
CINACALCET
+
4
+
C[C@@H](NCCCc1cccc(C(F)(F)F)c1)c1cccc2ccccc12
+
+
+
2
+
CHEMBL70418
+
CLOBAZAM
+
4
+
CN1C(=O)CC(=O)N(c2ccccc2)c2cc(Cl)ccc21
+
+
+
3
+
CHEMBL1628227
+
DOXEPIN
+
4
+
CN(C)CCC=C1c2ccccc2COc2ccccc21
+
+
+
4
+
CHEMBL1175
+
DULOXETINE
+
4
+
CNCC[C@H](Oc1cccc2ccccc12)c1cccs1
+
+
+
+
+
+
+
Initially, four categories of approved drugs are retrieved - the strong and moderate CYP3A4 inhibitors, and also the strong and moderate CYP2D6 inhibitors. CYP3A4 inhibitors are the largest cohort of all the cytochrome inhibitors known so far (based on clinical documentations).
+
+
+
+
Import and preprocess data
+
+
## When using pandas 2.2.2, numpy 2.0.0 and rdkit 2024.3.1
+# (all latest major versions at the time of writing,
+# note: rdkit has a latest minor release as 2024.03.4, which includes a patch for numpy 2.0)
+# Seems to work as a new df is generated but with error messages shown
+
+## Eventually using downgraded versions of pandas and numpy instead
+# pandas 2.1.4, numpy 1.26.4 & rdkit 2024.3.1 work with no error messages generated
+
+# preprocess canonical smiles
+from mol_prep import preprocess
+
+# cyp3a4 strong inhibitors
+df_3a4_s_inh = df_3a4_strong_inh.copy()
+df_3a4_s_inh_p = df_3a4_s_inh.apply(preprocess, axis=1)
+df_3a4_s_inh_p.head(3)
Here what I’m trying to do is to check structural validities of all the drug molecules, and one of the easiest things to do is to look at their chemical structures directly.
This is a small detour while checking structural validities due to the presence of two duplicated molecules, and since these two molecules consist of stereocentres, I’m just going to have a look at their stereochemistries.
+
+
+
+
quinidine
+
There are different stereochemistries spotted in the two quinidines shown below.
+
+
# Stereochem in RDKit
+# Older approach - AssignStereochemistry() -> this is used in datamol's standardize_mol(),
+# which is used in my small mol_prep.py script
+# Newer approach - FindPotentialStereo()
+
+
+
# Get 2D image of quinidine at row 3
+df_2d6_s_inh_p.loc[3, "rdkit_mol"]
+
+
+
+
+
+
+
# Get 2D image of quinidine at row 4
+df_2d6_s_inh_p.loc[4, "rdkit_mol"]
+
+
+
+
+
+
+
# Get SMILES for quinidine at row 3
+df_2d6_s_inh_p.loc[3, "canonical_smiles"]
# quinidine index row 4
+quinidine_4 = Chem.MolFromSmiles('C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2ccc(OC)cc12')
+rdCIPLabeler.AssignCIPLabels(quinidine_4)
+quinidine_4
+
+
+
+
+
+
Quinidine has 4 defined atom stereocentre count as per PubChem compound summary (as one of possible references for cross-checking) - this is based on the calculation for CHEMBL1294, which is the same as the quinidine spotted at index row 4. So I’m dropping the quinidine at index row 3 for now.
+
+
# Note: old index is unchanged for now (re-index later if needed)
+df_2d6_s_inh_p = df_2d6_s_inh_p.drop(labels =3)
+df_2d6_s_inh_p
+
+
+
+
+
+
+
+
+
chembl_id
+
pref_name
+
max_phase
+
canonical_smiles
+
rdkit_mol
+
standard_smiles
+
selfies
+
inchi
+
inchikey
+
+
+
+
+
0
+
CHEMBL894
+
BUPROPION
+
4.0
+
CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1
+
<rdkit.Chem.rdchem.Mol object at 0x135da3530>
+
CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1
+
[C][C][Branch1][#Branch2][N][C][Branch1][C][C]...
+
InChI=1S/C13H18ClNO/c1-9(15-13(2,3)4)12(16)10-...
+
SNPPWIUOZRMYNY-UHFFFAOYSA-N
+
+
+
1
+
CHEMBL41
+
FLUOXETINE
+
4.0
+
CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1
+
<rdkit.Chem.rdchem.Mol object at 0x135da3290>
+
CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1
+
[C][N][C][C][C][Branch2][Ring1][Ring2][O][C][=...
+
InChI=1S/C17H18F3NO/c1-21-12-11-16(13-5-3-2-4-...
+
RTHCYVBBDHJXIQ-UHFFFAOYSA-N
+
+
+
2
+
CHEMBL490
+
PAROXETINE
+
4.0
+
Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1
+
<rdkit.Chem.rdchem.Mol object at 0x135da3bc0>
+
Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1
+
[F][C][=C][C][=C][Branch2][Ring1][#Branch2][C@...
+
InChI=1S/C19H20FNO3/c20-15-3-1-13(2-4-15)17-7-...
+
AHOUBRCZNHFOSL-YOEHRIQHSA-N
+
+
+
4
+
CHEMBL1294
+
QUINIDINE
+
4.0
+
C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc...
+
<rdkit.Chem.rdchem.Mol object at 0x135da2d50>
+
C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc...
+
[C][=C][C@H1][C][N][C][C][C@H1][Ring1][=Branch...
+
InChI=1S/C20H24N2O2/c1-3-13-12-22-9-7-14(13)10...
+
LOUPRKONTZGTKE-LHHVKLHASA-N
+
+
+
+
+
+
+
+
+
+
itraconazole
+
Two itraconazoles are also found with different stereochemistries.
+
+
# Get SMILES of itraconazole at index row 4
+df_3a4_s_inh_p.loc[4, "canonical_smiles"]
Clearly, even if the SMILES of these two itraconzoles are not converted into RDKit molecules, we can probably tell one of them has stereochemistries and the other one is without due to the presence of “@” in the SMILES string for the one at index row 4. The output images show exactly that - one with chiral centres, where the other one doesn’t have any.
+
PubChem calculations have however generated different result for itraconazole. It seems it only has one defined atom stereocentre count and two undefined stereocentre counts (PubChem reference).
+
I’ve also noted that the two itraconzoles obtained from ChEMBL have different ChEMBL ID numbers (ChEMBL IDs: CHEMBL22587 and CHEMBL64391) to the one calculated in PubChem (ChEMBL ID: CHEMBL224725). So below I’ve looked into CHEMBL224725 first.
+
Then I realise that if I search for “itraconazole” directly in ChEMBL, only four entries will appear with ChEMBL IDs of CHEMBL64391, CHEMBL22587, CHEMBL882 and CHEMBL5090785, and there is no ChEMBL224725. This is all due to a small spelling error (which is most likely a typo by accident) of itraconazole - being spelled as “itraconzole”, which is also carried over into PubChem as well. I have checked again to make sure both “itraconzole” and the usual itraconazole are referring to the same chemical structure. Below are some screenshots showing the typo.
+
So to add this likely-mis-spelled “itraconzole” into the dataframe, I literally just add it into the SQL query above when obtaining drug information through chembl_downloader.
+
+
# SMILES of new addition - "itraconzole"
+df_3a4_s_inh_p.loc[6, "canonical_smiles"]
# Labelling stereocentres of new addition - "itraconzole"
+itracon_6 = Chem.MolFromSmiles("CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5COC(Cn6cncn6)(c6ccc(F)cc6F)O5)cc4)CC3)cc2)c1=O")
+rdCIPLabeler.AssignCIPLabels(itracon_6)
+itracon_6
+
+
+
+
+
+
There is only one stereocentre for “itraconzole”, which will match the CHEMBL224725 entry for “itraconzole” in PubChem. Without looking into other cross-referencing sources, and if only sticking with PubChem for now, I’ve then gone back to check all 3 (stereochemically-)different versions of itraconazole and found that the RDKit stereochemical calculations of these 3 itraconazoles have all basically matched their equivalent PubChem computations for atom sterecentre counts.
Dataframe df_3a4_s_inh_p (the preprocessed strong CYP3A4 inhibitors) containing 3 different itraconazoles is then updated below to remove two of the triplicated entries.
+
+
# Preprocessed df of strong cyp3a4 inhibitors
+df_3a4_s_inh_p.head(10)
+
+
+
+
+
+
+
+
+
chembl_id
+
pref_name
+
max_phase
+
canonical_smiles
+
rdkit_mol
+
standard_smiles
+
selfies
+
inchi
+
inchikey
+
+
+
+
+
0
+
CHEMBL2403108
+
CERITINIB
+
4.0
+
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
+
<rdkit.Chem.rdchem.Mol object at 0x135da19a0>
+
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
+
[C][C][=C][C][Branch2][Ring2][=Branch1][N][C][...
+
InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-...
+
VERWOWGGCGHDQE-UHFFFAOYSA-N
+
+
+
1
+
CHEMBL1741
+
CLARITHROMYCIN
+
4.0
+
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1b60>
+
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
+
[C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran...
+
InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21...
+
AGOYDEPGAOXOCK-KCBOHYOISA-N
+
+
+
2
+
CHEMBL2216870
+
IDELALISIB
+
4.0
+
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1a80>
+
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
+
[C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][...
+
InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-...
+
IFSDAJWBUCMOAH-HNNXBMFYSA-N
+
+
+
3
+
CHEMBL115
+
INDINAVIR
+
4.0
+
CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1ee0>
+
CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](...
+
[C][C][Branch1][C][C][Branch1][C][C][N][C][=Br...
+
InChI=1S/C36H47N5O4/c1-36(2,3)39-35(45)31-24-4...
+
CBVCZFGXHXORBI-PXQQMZJSSA-N
+
+
+
4
+
CHEMBL22587
+
ITRACONAZOLE
+
NaN
+
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5CO[C@](...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1cb0>
+
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5CO[C@](...
+
[C][C][C][Branch1][C][C][N][N][=C][N][Branch2]...
+
InChI=1S/C35H38Cl2N8O4/c1-3-25(2)45-34(46)44(2...
+
VHVPQPYKVGDNFY-ZPGVKDDISA-N
+
+
+
5
+
CHEMBL64391
+
ITRACONAZOLE
+
4.0
+
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1d90>
+
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn...
+
[C][C][C][Branch1][C][C][N][N][=C][N][Branch2]...
+
InChI=1S/C35H38Cl2N8O4/c1-3-25(2)45-34(46)44(2...
+
VHVPQPYKVGDNFY-UHFFFAOYSA-N
+
+
+
6
+
CHEMBL224725
+
ITRACONZOLE
+
NaN
+
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5COC(Cn6...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1f50>
+
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OC[C@H]5COC(Cn6...
+
[C][C][C][Branch1][C][C][N][N][=C][N][Branch2]...
+
InChI=1S/C35H38F2N8O4/c1-3-25(2)45-34(46)44(24...
+
HUADITLKOCMHSB-RPOYNCMSSA-N
+
+
+
7
+
CHEMBL157101
+
KETOCONAZOLE
+
4.0
+
CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c...
+
<rdkit.Chem.rdchem.Mol object at 0x135da2030>
+
CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c...
+
[C][C][=Branch1][C][=O][N][C][C][N][Branch2][R...
+
InChI=1S/C26H28Cl2N4O4/c1-19(33)31-10-12-32(13...
+
XMAYWYJOQHXEEK-UHFFFAOYSA-N
+
+
+
8
+
CHEMBL45816
+
MIBEFRADIL
+
4.0
+
COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1e70>
+
COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c...
+
[C][O][C][C][=Branch1][C][=O][O][C@][Branch2][...
+
InChI=1S/C29H38FN3O3/c1-20(2)28-23-12-11-22(30...
+
HBNPJJILLOYFJU-VMPREFPWSA-N
+
+
+
9
+
CHEMBL623
+
NEFAZODONE
+
4.0
+
CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c...
+
<rdkit.Chem.rdchem.Mol object at 0x135da20a0>
+
CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c...
+
[C][C][C][=N][N][Branch2][Ring1][=Branch2][C][...
+
InChI=1S/C25H32ClN5O2/c1-2-24-27-31(25(32)30(2...
+
VRBKIVRKKCLPHA-UHFFFAOYSA-N
+
+
+
+
+
+
+
I’m keeping the one with max phase marked as 4.0 (due to the other two having “NaN” with no relevant medical or therapeutic indications data documented in PubChem).
+
+
# Note old index unchanged (re-index later if needed)
+# Dropping itraconazole at index rows 4 & 6
+df_3a4_s_inh_p = df_3a4_s_inh_p.drop(labels = [4, 6])
+df_3a4_s_inh_p.head(10)
+
+
+
+
+
+
+
+
+
chembl_id
+
pref_name
+
max_phase
+
canonical_smiles
+
rdkit_mol
+
standard_smiles
+
selfies
+
inchi
+
inchikey
+
+
+
+
+
0
+
CHEMBL2403108
+
CERITINIB
+
4.0
+
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
+
<rdkit.Chem.rdchem.Mol object at 0x135da19a0>
+
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
+
[C][C][=C][C][Branch2][Ring2][=Branch1][N][C][...
+
InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-...
+
VERWOWGGCGHDQE-UHFFFAOYSA-N
+
+
+
1
+
CHEMBL1741
+
CLARITHROMYCIN
+
4.0
+
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1b60>
+
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
+
[C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran...
+
InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21...
+
AGOYDEPGAOXOCK-KCBOHYOISA-N
+
+
+
2
+
CHEMBL2216870
+
IDELALISIB
+
4.0
+
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1a80>
+
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
+
[C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][...
+
InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-...
+
IFSDAJWBUCMOAH-HNNXBMFYSA-N
+
+
+
3
+
CHEMBL115
+
INDINAVIR
+
4.0
+
CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1ee0>
+
CC(C)(C)NC(=O)[C@@H]1CN(Cc2cccnc2)CCN1C[C@@H](...
+
[C][C][Branch1][C][C][Branch1][C][C][N][C][=Br...
+
InChI=1S/C36H47N5O4/c1-36(2,3)39-35(45)31-24-4...
+
CBVCZFGXHXORBI-PXQQMZJSSA-N
+
+
+
5
+
CHEMBL64391
+
ITRACONAZOLE
+
4.0
+
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1d90>
+
CCC(C)n1ncn(-c2ccc(N3CCN(c4ccc(OCC5COC(Cn6cncn...
+
[C][C][C][Branch1][C][C][N][N][=C][N][Branch2]...
+
InChI=1S/C35H38Cl2N8O4/c1-3-25(2)45-34(46)44(2...
+
VHVPQPYKVGDNFY-UHFFFAOYSA-N
+
+
+
7
+
CHEMBL157101
+
KETOCONAZOLE
+
4.0
+
CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c...
+
<rdkit.Chem.rdchem.Mol object at 0x135da2030>
+
CC(=O)N1CCN(c2ccc(OCC3COC(Cn4ccnc4)(c4ccc(Cl)c...
+
[C][C][=Branch1][C][=O][N][C][C][N][Branch2][R...
+
InChI=1S/C26H28Cl2N4O4/c1-19(33)31-10-12-32(13...
+
XMAYWYJOQHXEEK-UHFFFAOYSA-N
+
+
+
8
+
CHEMBL45816
+
MIBEFRADIL
+
4.0
+
COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1e70>
+
COCC(=O)O[C@]1(CCN(C)CCCc2nc3ccccc3[nH]2)CCc2c...
+
[C][O][C][C][=Branch1][C][=O][O][C@][Branch2][...
+
InChI=1S/C29H38FN3O3/c1-20(2)28-23-12-11-22(30...
+
HBNPJJILLOYFJU-VMPREFPWSA-N
+
+
+
9
+
CHEMBL623
+
NEFAZODONE
+
4.0
+
CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c...
+
<rdkit.Chem.rdchem.Mol object at 0x135da20a0>
+
CCc1nn(CCCN2CCN(c3cccc(Cl)c3)CC2)c(=O)n1CCOc1c...
+
[C][C][C][=N][N][Branch2][Ring1][=Branch2][C][...
+
InChI=1S/C25H32ClN5O2/c1-2-24-27-31(25(32)30(2...
+
VRBKIVRKKCLPHA-UHFFFAOYSA-N
+
+
+
10
+
CHEMBL584
+
NELFINAVIR
+
4.0
+
Cc1c(O)cccc1C(=O)N[C@@H](CSc1ccccc1)[C@H](O)CN...
+
<rdkit.Chem.rdchem.Mol object at 0x135da2180>
+
Cc1c(O)cccc1C(=O)N[C@@H](CSc1ccccc1)[C@H](O)CN...
+
[C][C][=C][Branch1][C][O][C][=C][C][=C][Ring1]...
+
InChI=1S/C32H45N3O4S/c1-21-25(15-10-16-28(21)3...
+
QAGYKUNXZHXKMR-HKWSIXNMSA-N
+
+
+
11
+
CHEMBL3545110
+
RIBOCICLIB
+
4.0
+
CN(C)C(=O)c1cc2cnc(Nc3ccc(N4CCNCC4)cn3)nc2n1C1...
+
<rdkit.Chem.rdchem.Mol object at 0x135da22d0>
+
CN(C)C(=O)c1cc2cnc(Nc3ccc(N4CCNCC4)cn3)nc2n1C1...
+
[C][N][Branch1][C][C][C][=Branch1][C][=O][C][=...
+
InChI=1S/C23H30N8O/c1-29(2)22(32)19-13-16-14-2...
+
RHXHGRAEPCAFML-UHFFFAOYSA-N
+
+
+
+
+
+
+
After cleaning up the duplicated structures, below are the full sets of strong and moderate CYP3A4 inhibitors for structural checking.
MCS is something I’m interested in trying out so below are some examples of finding MCS in these CYP inhibitors. Please note that MCS may not be the most suitable strategy to look at these CYP inhibitors, I’m only using it to become a bit more familiar with it so that I can better understand MCS.
+
Some information regarding MCS in RDKit:
+
+
FindMCS is for 2 or more molecules and returns single-fragment MCS - based on FMCS algorithm (Dalke and Hastings 2013)
+
RascalMCES (maximum common edge substructures) is for 2 molecules only and returns multi-fragment MCES. A RDKit blog post by Dave Cosgrove talks about this in more details
I’m starting with the strong CYP3A4 inhibitors first.
+
+
# Get list of RDKit mols
+mols_s3a4 =list(df_3a4_s_inh_p["rdkit_mol"])
+
+# Find MCS in mols
+s3a4_mcs = rdFMCS.FindMCS(mols_s3a4)
+
+# Get images of highlighted MCS for strong CYP3A4 inhibitors
+Draw.MolsToGridImage(
+ mols_s3a4,
+ subImgSize=(400, 300),
+ molsPerRow=2,
+ legends =list(df_3a4_s_inh_p["pref_name"]),
+ highlightAtomLists=[m.GetSubstructMatch(s3a4_mcs.queryMol) for m in mols_s3a4]
+ )
+
+
+
+
+
You can get the number of atoms and bonds and also SMARTS string for the MCS like this below.
One way to customise MCS is via reducing molecule threshold to relax the MCS rule as suggested by the TeachOpenCADD reference above.
+
+
s3a4_mcs_80 = rdFMCS.FindMCS(mols_s3a4, threshold=0.8)
+
+Draw.MolsToGridImage(
+ mols_s3a4,
+ subImgSize=(400, 300),
+ molsPerRow=2,
+ legends =list(df_3a4_s_inh_p["pref_name"]),
+ highlightAtomLists=[m.GetSubstructMatch(s3a4_mcs_80.queryMol) for m in mols_s3a4]
+ )
+
+
+
+
+
+
# Without changing threshold
+s3a4_mcs1 = Chem.MolFromSmarts(s3a4_mcs.smartsString)
+
+# Lowered MCS threshold to 80% of mols
+s3a4_mcs2 = Chem.MolFromSmarts(s3a4_mcs_80.smartsString)
+
+Draw.MolsToGridImage([s3a4_mcs1, s3a4_mcs2], legends=["MCS1", "MCS2 with threshold = 0.8"])
+
+
+
+
+
Here the MCS differs between different MCS thresholds - when the threshold is not changed, it shows a partial contour of a ring structure, whereas the lowered threshold shows more of an alkyl chain structure.
+
+
+
+
Moderate CYP3A4 inhibitors
+
This is then followed by the moderate inhibitors for CYP3A4.
+
+
# Get list of RDKit mols
+mols_m3a4 =list(df_3a4_m_inh_p["rdkit_mol"])
+
+# Find MCS in mols
+m3a4_mcs = rdFMCS.FindMCS(mols_m3a4)
+
+# Get images of highlighted MCS for moderate CYP3A4 inhibitors
+Draw.MolsToGridImage(
+ mols_m3a4,
+ subImgSize=(400, 300),
+ molsPerRow=2,
+ legends =list(df_3a4_s_inh_p["pref_name"]),
+ highlightAtomLists=[m.GetSubstructMatch(m3a4_mcs.queryMol) for m in mols_m3a4]
+ )
+
+
+
+
+
Aromatic (and macrolide) rings are highlighted in the MCS for this group.
+
+
+
+
All CYP3A4 inhibitors
+
The moderate CYP3A4 inhibitors are then added next in order to see if MCS changes when looking at all of the CYP3A4 inhibitors.
+
+
# Concatenate dfs for moderate & strong CYP3A4 inhibitors
+df_3a4_all = pd.concat([df_3a4_s_inh_p, df_3a4_m_inh_p])
+# index un-changed for now
+print(df_3a4_all.shape)
+df_3a4_all.head(3)
+
+
(27, 9)
+
+
+
+
+
+
+
+
+
+
chembl_id
+
pref_name
+
max_phase
+
canonical_smiles
+
rdkit_mol
+
standard_smiles
+
selfies
+
inchi
+
inchikey
+
+
+
+
+
0
+
CHEMBL2403108
+
CERITINIB
+
4.0
+
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
+
<rdkit.Chem.rdchem.Mol object at 0x135da19a0>
+
Cc1cc(Nc2ncc(Cl)c(Nc3ccccc3S(=O)(=O)C(C)C)n2)c...
+
[C][C][=C][C][Branch2][Ring2][=Branch1][N][C][...
+
InChI=1S/C28H36ClN5O3S/c1-17(2)37-25-15-21(20-...
+
VERWOWGGCGHDQE-UHFFFAOYSA-N
+
+
+
1
+
CHEMBL1741
+
CLARITHROMYCIN
+
4.0
+
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1b60>
+
CC[C@H]1OC(=O)[C@H](C)[C@@H](O[C@H]2C[C@@](C)(...
+
[C][C][C@H1][O][C][=Branch1][C][=O][C@H1][Bran...
+
InChI=1S/C38H69NO13/c1-15-26-38(10,45)31(42)21...
+
AGOYDEPGAOXOCK-KCBOHYOISA-N
+
+
+
2
+
CHEMBL2216870
+
IDELALISIB
+
4.0
+
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
+
<rdkit.Chem.rdchem.Mol object at 0x135da1a80>
+
CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n...
+
[C][C][C@H1][Branch1][#C][N][C][=N][C][=N][C][...
+
InChI=1S/C22H18FN7O/c1-2-15(28-20-18-19(25-11-...
+
IFSDAJWBUCMOAH-HNNXBMFYSA-N
+
+
+
+
+
+
+
+
mols_3a4_all =list(df_3a4_all["rdkit_mol"])
+
+# Find MCS for all CYP3A4 inhibitors
+all_3a4_mcs = rdFMCS.FindMCS(mols_3a4_all)
+
+# All CYP3A4 inhibitors
+Draw.MolsToGridImage(
+ mols_3a4_all,
+ subImgSize=(400, 300),
+ molsPerRow=2,
+ legends =list(df_3a4_all["pref_name"]),
+ highlightAtomLists=[m.GetSubstructMatch(all_3a4_mcs.queryMol) for m in mols_3a4_all]
+ )
+
+
+
+
+
It appears the MCS for most of them involves (partial) rings (e.g. cycloalkane, aromatic and macrolide ones). This result is basically not very different from when we’ve looked at the the CYP3A4 inhibitors separately in their moderate and strong potencies. The next thing I want to try is to add in the ring bonds matching.
+
+
# matching ring bonds
+all_3a4_mcs_ring = rdFMCS.FindMCS(mols_3a4_all, ringMatchesRingOnly=True)
+
+Draw.MolsToGridImage(
+ mols_3a4_all,
+ subImgSize=(400, 300),
+ molsPerRow=2,
+ legends =list(df_3a4_all["pref_name"]),
+ highlightAtomLists=[m.GetSubstructMatch(all_3a4_mcs_ring.queryMol) for m in mols_3a4_all]
+ )
+
+
+
+
+
The ring bond matching also shows a very similar result here as it only restricts the MCS matching to ring bonds only.
+
+
Some other interesting code re. MCS
+
One of the code examples I’d like to try next, in order to see if it makes any differences from above, is the code snippets shared by a RDKit post from Paolo Tosco - “New MCS features in 2023.09.1” - the “Custom Python AtomCompare and BondCompare classes” section.
+
Some notes from me:
+
+
AtomCompare & BondCompare - looking at elements/bond orders/aromaticities in ring systems or
+
custom subclasses in AtomCompare & BondCompare - looking at elements/bond orders/aromaticities in non-ring systems
+
+
i.e. customise parameters using rdFMCS.MCSParameters()
+
I’ll also attempt to add some code comments below to explain how the code works (anyone’s welcomed to report any issues or changes for this part).
+
+
Ring systems
+
Here, I’m trying the AtomCompare & BondCompare along with RingMatchesRingOnly and CompleteRingsOnly first.
+
+
+Code
+
## Customise MCS parameters
+# Initiate a MCSParameter object
+params = rdFMCS.MCSParameters()
+# Define atom typer to be used to compare elements within rdFMCS.AtomCompare class
+params.AtomTyper = rdFMCS.AtomCompare.CompareElements
+# Define bond typer to be used to compare bond orders within rdFMCS.BondCompare class
+params.BondTyper = rdFMCS.BondCompare.CompareOrder
+# RingMatchesRingOnly - ring atoms to match other ring atoms only
+# CompleteRingsOnly - match full rings only
+params.BondCompareParameters.RingMatchesRingOnly =True
+params.BondCompareParameters.CompleteRingsOnly =False
+
+all_3a4_ringMCS = rdFMCS.FindMCS(mols_3a4_all, params)
+
+Draw.MolsToGridImage(
+ mols_3a4_all,
+ subImgSize=(400, 300),
+ molsPerRow=3,
+ legends =list(df_3a4_all["pref_name"]),
+ highlightAtomLists=[m.GetSubstructMatch(all_3a4_ringMCS.queryMol) for m in mols_3a4_all]
+ )
+
+
+
+
+
+
Some MCS characteristics noted after trying this:
+
+
When both BondCompareParameters are set to true, no rings are highlighted (apart from ethyl chains being highlighted in every molecule)
+
When turning off CompleteRingsOnly, this allows partial rings to be shown in highlighted MCS
+
A similar output is generated when using ringMatchesRingOnly = True in FindMCS() earlier for all CYP3A4 inhibitors
+
+
+
Non-ring systems
+
The class code below is also borrowed from the RDKit blog post, which explains why it’s done as a custom “class” code and not a “function” one.
+
+
+Code
+
# I've had to think harder about the what the class code are doing below,
+# especially the differences between comparing bond orders and ring atoms...
+# I can only describe it as both a restrictive (matching non-ring bonds only)
+# and lenient (but still comparing ring atoms) process in order to cover the non-ring parts (?)
+
+# Compare bond orders outside ring systems using rdFMCS.MCSBondCompare
+# Using class code that will call a function object
+class CompareOrderOutsideRings(rdFMCS.MCSBondCompare):
+
+def__call__(self, p, mol1, bond1, mol2, bond2):
+
+# Get bonds 1 and 2 based on bond indices for mols 1 and 2
+ b1 = mol1.GetBondWithIdx(bond1)
+ b2 = mol2.GetBondWithIdx(bond2)
+# If bonds 1 and 2 are both in ring or if their bond types are the same
+if (b1.IsInRing() and b2.IsInRing()) or (b1.GetBondType() == b2.GetBondType()):
+# If using stereo matching parameter and not checking bond stereo descriptors,
+# return bonds that are not using stereo matching parameter
+if (p.MatchStereo andnotself.CheckBondStereo(p, mol1, bond1, mol2, bond2)):
+returnFalse
+# If using ring bonds-matching-ring bonds parameter
+# return mols/bonds that are part of a ring only
+if p.RingMatchesRingOnly:
+returnself.CheckBondRingMatch(p, mol1, bond1, mol2, bond2)
+returnTrue
+# Only match bonds that are not part of ring systems
+returnFalse
+
+# Compare atom elements outside ring systems using rdFMCS.MCSAtomCompare
+class CompareElementsOutsideRings(rdFMCS.MCSAtomCompare):
+
+def__call__(self, p, mol1, atom1, mol2, atom2):
+
+# Get atoms 1 and 2 based on atom indices for mols 1 and 2
+ a1 = mol1.GetAtomWithIdx(atom1)
+ a2 = mol2.GetAtomWithIdx(atom2)
+# If atomic numbers for both atoms are different and the atoms are not in ring systems,
+# return atoms that instead have the same atomic numbers and in rings systems
+if (a1.GetAtomicNum() != a2.GetAtomicNum()) andnot (a1.IsInRing() and a2.IsInRing()):
+returnFalse
+# If using matching atom chirality parameter and not checking both atoms have a chiral tag,
+# return atoms that are not using the matching atom chirality parameter
+if (p.MatchChiralTag andnotself.CheckAtomChirality(p, mol1, atom1, mol2, atom2)):
+returnFalse
+# If using ring bonds-matching-ring bonds parameter,
+# return mols/atoms that are part of a ring only
+if p.RingMatchesRingOnly:
+returnself.CheckAtomRingMatch(p, mol1, atom1, mol2, atom2)
+# Only match ring atoms against ring atoms
+returnTrue
+
+params_or = rdFMCS.MCSParameters()
+params_or.AtomTyper = CompareElementsOutsideRings()
+params_or.BondTyper = CompareOrderOutsideRings()
+params_or.BondCompareParameters.RingMatchesRingOnly =True
+params_or.BondCompareParameters.CompleteRingsOnly =True
+
+all_3a4_orMCS = rdFMCS.FindMCS(mols_3a4_all, params_or)
+
+Draw.MolsToGridImage(
+ mols_3a4_all,
+ subImgSize=(500, 400),
+ molsPerRow=3,
+ legends =list(df_3a4_all["pref_name"]),
+ highlightAtomLists=[m.GetSubstructMatch(all_3a4_orMCS.queryMol) for m in mols_3a4_all]
+ )
+
+
+
+
+
+
By using the suggested class code above, the MCS has indeed become larger, where a full ring is highlighted as the MCS.
+
A second code example from iwatobipen is to highlight molecular differences, which is different from highlighting only the MCSes. Alternative URL link and more examples can be accessed via RDKit Cookbook. Possible use cases of this code may be when dealing with a large set of analogues with changes in R-groups or during large compound screening and search (just as a few examples only). The main thing I can see from the code is that it begins with finding MCS for the input molecules, then uses SMARTS strings of the MCS in order to find atoms not within the MCS (using GetSubstructMatch() part) then highlights that part of the molecules.
+
+
+
+
All CYP2D6 inhibitors
+
Because of how the MCSes have turned out for CYP3A4 inhibitors above, I think I should just look at CYP2D6 inhibitors as a whole here. First thing is to combine the dataframes between the moderate and strong inhibitor groups.
+
+
# Concatenate dfs
+df_2d6_all = pd.concat([df_2d6_s_inh_p, df_2d6_m_inh_p])
+# index un-changed for now
+print(df_2d6_all.shape)
+df_2d6_all.head()
+
+
(14, 9)
+
+
+
+
+
+
+
+
+
+
chembl_id
+
pref_name
+
max_phase
+
canonical_smiles
+
rdkit_mol
+
standard_smiles
+
selfies
+
inchi
+
inchikey
+
+
+
+
+
0
+
CHEMBL894
+
BUPROPION
+
4.0
+
CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1
+
<rdkit.Chem.rdchem.Mol object at 0x135da3530>
+
CC(NC(C)(C)C)C(=O)c1cccc(Cl)c1
+
[C][C][Branch1][#Branch2][N][C][Branch1][C][C]...
+
InChI=1S/C13H18ClNO/c1-9(15-13(2,3)4)12(16)10-...
+
SNPPWIUOZRMYNY-UHFFFAOYSA-N
+
+
+
1
+
CHEMBL41
+
FLUOXETINE
+
4.0
+
CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1
+
<rdkit.Chem.rdchem.Mol object at 0x135da3290>
+
CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1
+
[C][N][C][C][C][Branch2][Ring1][Ring2][O][C][=...
+
InChI=1S/C17H18F3NO/c1-21-12-11-16(13-5-3-2-4-...
+
RTHCYVBBDHJXIQ-UHFFFAOYSA-N
+
+
+
2
+
CHEMBL490
+
PAROXETINE
+
4.0
+
Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1
+
<rdkit.Chem.rdchem.Mol object at 0x135da3bc0>
+
Fc1ccc([C@@H]2CCNC[C@H]2COc2ccc3c(c2)OCO3)cc1
+
[F][C][=C][C][=C][Branch2][Ring1][#Branch2][C@...
+
InChI=1S/C19H20FNO3/c20-15-3-1-13(2-4-15)17-7-...
+
AHOUBRCZNHFOSL-YOEHRIQHSA-N
+
+
+
4
+
CHEMBL1294
+
QUINIDINE
+
4.0
+
C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc...
+
<rdkit.Chem.rdchem.Mol object at 0x135da2d50>
+
C=C[C@H]1CN2CC[C@H]1C[C@@H]2[C@@H](O)c1ccnc2cc...
+
[C][=C][C@H1][C][N][C][C][C@H1][Ring1][=Branch...
+
InChI=1S/C20H24N2O2/c1-3-13-12-22-9-7-14(13)10...
+
LOUPRKONTZGTKE-LHHVKLHASA-N
+
+
+
0
+
CHEMBL254328
+
ABIRATERONE
+
4.0
+
C[C@]12CC[C@H](O)CC1=CC[C@@H]1[C@@H]2CC[C@]2(C...
+
<rdkit.Chem.rdchem.Mol object at 0x135dd0270>
+
C[C@]12CC[C@H](O)CC1=CC[C@@H]1[C@@H]2CC[C@]2(C...
+
[C][C@][C][C][C@H1][Branch1][C][O][C][C][Ring1...
+
InChI=1S/C24H31NO/c1-23-11-9-18(26)14-17(23)5-...
+
GZOSMCIZMLWJML-VJLLXTKPSA-N
+
+
+
+
+
+
+
+
mols_all_2d6 =list(df_2d6_all["rdkit_mol"])
+
+# Find MCS for all CYP2D6 inhibitors
+all_2d6_mcs = rdFMCS.FindMCS(mols_all_2d6)
+
+# Get images of highlighted MCS for all CYP2D6 inhibitors
+Draw.MolsToGridImage(
+ mols_all_2d6,
+ subImgSize=(400, 300),
+ molsPerRow=2,
+ legends =list(df_2d6_all["pref_name"]),
+ highlightAtomLists=[m.GetSubstructMatch(all_2d6_mcs.queryMol) for m in mols_all_2d6]
+ )
+
+
+
+
+
Again, only a phenyl ring is highlighted as the MCS so this is not informative at all.
+
+
+
+
+
Some findings and future work
+
Below are some of my small findings and thoughts.
+
+
As mentioned at the very top in the outline section, rings (heterocycles, aromatic or fused ones) are everywhere in the MCSes for CYP3A4 and CYP2D6 inhibitors, and they are very common in known approved drugs
+
Looking at CYP structures in relation to these inhibitors may be more meaningful and also may reveal more insights about possible binding sites or mechanisms of actions of CYP inhibitions for these compounds. This may also be further explored in parallel to the actual drug targets of these CYP inhibitors e.g. binding site on CYP isoenzyme versus binding site on drug target protein as there are different classes of drugs within these CYP inhibitors. For example, some of CYP2D6 inhibitors are drugs acting on central nervous system (about 9 out of 14 drugs) - e.g. bupropion, fluoxetine, paroxetine, clobazam, doxepin, duloxetine, locaserin, moclobemide and rolapitant. For the CYP3A4 inhibitors, they are in several different therapeutic classes e.g. antivirals, antifungals, antibacterials, kinase inhibitors etc.
+
It may be a bit more interesting to compare the MCSes between CYP3A4 and CYP2D6 substrates (adding in other substrates for other CYPs)
+
Future posts may involve looking at CYP substrates using different cheminformatics strategies or doing molecular docking within a notebook setting (has this been done before?)
+
CYP inducers are a different story as they tend to increase drug metabolisms via CYP inductions, which are more likely to do with losing therapeutic effects than gaining adverse effects, so they may be looked at further down the line
+
MCS may not be absolutely useful in every scenario as I think it aims to look for the most maximum common substructures within a set of molecules, so not every molecule will be able to have a MCS shown (e.g. in a very diverse chemical set), other similarity searching techniques should probably be used instead if needed
+
+
+
+
+
Acknowledgements
+
Thanks to every contributor, developer or authors of every software package used in this post, and also the online communities behind them. Before I forget, the thanks would also be extended to the authors of the journal papers cited in this post. Lastly, special thanks to Noel O’Boyle for being very patient with reading my awfully-long old draft earlier and pointed out some useful things to note and change (I kind of got lost when writing the draft… due to it being a “free-style post”, I should avoid doing this again).
+
+
+
+
+
+
References
+
+Curran, Mark E, Igor Splawski, Katherine W Timothy, G.Michael Vincen, Eric D Green, and Mark T Keating. 1995. “A Molecular Basis for Cardiac Arrhythmia: HERG Mutations Cause Long QT Syndrome.”Cell 80 (5): 795–803. https://doi.org/10.1016/0092-8674(95)90358-5.
+
+
+Dalke, Andrew, and Janna Hastings. 2013. “FMCS: A Novel Algorithm for the Multiple MCS Problem.”Journal of Cheminformatics 5 (S1). https://doi.org/10.1186/1758-2946-5-s1-o6.
+
+
+Doveston, Richard G., Paolo Tosatti, Mark Dow, Daniel J. Foley, Ho Yin Li, Amanda J. Campbell, David House, Ian Churcher, Stephen P. Marsden, and Adam Nelson. 2015. “A Unified Lead-Oriented Synthesis of over Fifty Molecular Scaffolds.”Organic & Biomolecular Chemistry 13 (3): 859–65. https://doi.org/10.1039/c4ob02287d.
+
+Gaulton, Anna, Anne Hersey, Michał Nowotka, A. Patrícia Bento, Jon Chambers, David Mendez, Prudence Mutowo, et al. 2016. “The ChEMBL database in 2017.”Nucleic Acids Research 45 (D1): D945–54. https://doi.org/10.1093/nar/gkw1074.
+
+
+Guengerich, F. Peter. 2020. “A History of the Roles of Cytochrome P450 Enzymes in the Toxicity of Drugs.”Toxicological Research 37 (1): 1–23. https://doi.org/10.1007/s43188-020-00056-z.
+
+
+Jadhav, Ajit, Rafaela S. Ferreira, Carleen Klumpp, Bryan T. Mott, Christopher P. Austin, James Inglese, Craig J. Thomas, David J. Maloney, Brian K. Shoichet, and Anton Simeonov. 2010. “Quantitative Analyses of Aggregation, Autofluorescence, and Reactivity Artifacts in a Screen for Inhibitors of a Thiol Protease.”Journal of Medicinal Chemistry 53 (1): 37–51. https://doi.org/10.1021/jm901070c.
+
+
+Taylor, Richard D., Malcolm MacCoss, and Alastair D. G. Lawson. 2014. “Rings in Drugs.”Journal of Medicinal Chemistry 57 (14): 5845–59. https://doi.org/10.1021/jm4017625.
+
+
]]>
+ Metabolism
+ Toxicology
+ Structural alerts
+ Pandas
+ RDKit
+ ChEMBL database
+ Python
+ https://jhylin.github.io/Data_in_life_blog/posts/20_Cyp3a4_2d6_inh/1_CYP450_drugs.html
+ Wed, 21 Aug 2024 12:00:00 GMT
+
+
+
+ Boosted trees
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/19_ML2-3_Boosted_trees/1_adaboost_xgb.html
+
+
Some introductions
+
I’ve somehow promised myself to do a tree series on machine learning and glad I’ve made it to the boosted trees part (it took a while…). This is also likely my last post on this topic for now as there are other things I want to explore in the near future. Hopefully this is somewhat useful for anyone who’s new to this.
+
+
+
AdaBoost
+
Adaptive Boost or AdaBoost has originated from Robert E. Schapire in 1990 (Schapire 1990), (Raschka, Liu, and Mirjalili 2022), and was further introduced in 1996 by Robert Schapire and Yoav Freund at a conference which also led to a publication (Freund and Schapire 1997).
+
As quoted from scikit-learn, an AdaBoost algorithm is doing this:
+
+
…fit a sequence of weak learners (i.e., models that are only slightly better than random guessing, such as small decision trees) on repeatedly modified versions of the data.
+
+
+
+
+
+
+
+Note
+
+
+
+
Weak learner means an ensemble of very simple base classifiers such as decision tree stumps (Raschka, Liu, and Mirjalili 2022)
+
+
+
During the process of running the algorithm, increased weights are given to the incorrectly predicted samples at each iteration, and less weights are given to the correctly predicted ones. This then forces the AdaBoost models to focus more on the less accurately predicted samples with the aim to improve ensemble performance. The predictions from these iterations are combined to produce a final prediction via a weighted majority vote style, which is a well-known signature of tree models. Overall, AdaBoost algorithm can be used for classification or regression problems. The main difference between bagging and boosting is that boosting only uses random subsets of training samples drawn from the training dataset without any replacements (Raschka, Liu, and Mirjalili 2022). One caveat to note is that AdaBoost tend to overfit training data (high variance).
+
Parameters to tune:
+
+
n_estimators - number of weak learners
+
learning_rate - contributions of weak learners in the final combination
+
max_depth - depth of trees
+
min_samples_split - minimum required number of samples to consider a split
+
+
+
+
+
Gradient boosted trees
+
Essentially a similar concept is behind gradient boosted trees where a series of weak learners is trained in order to create a stronger ensemble of models (Raschka, Liu, and Mirjalili 2022). However, some differences between these two types of boosted trees (e.g. AdaBoost and XGBoost) should be noted, and rather than describing them in a paragraph, I’ve summarised them in a table below.
+
+
+
+
+
+
+
AdaBoost
+
XGBoost
+
+
+
+
+
trains weak learners based on errors from previous decision tree stump
+
trains weak learners that are deeper than decision tree stumps with a max depth of 3 to 6 (or max number of leaf nodes from 8 to 64)
+
+
+
uses prediction errors to calculate sample weights and classifier weights
+
uses prediction errors directly to produce the target variable to fit the next tree
+
+
+
uses individual weighting terms for each tree
+
uses a global learning rate for each tree
+
+
+
+
Differences between XGBoost and AdaBoost (Raschka, Liu, and Mirjalili 2022)
+
+
XGBoost or extreme gradient boosting (Chen and Guestrin 2016) is one of the most commonly used open-source packages, originally developed at the University of Washington by T. Chen and C. Guestrin, that uses stochastic gradient boosting to build an ensemble of predictive models.
Main parameters to tune as suggested by (Bruce, Bruce, and Gedeck 2020):
+
+
subsample - controls fraction of observations that should be sampled at each iteration or a subsample ratio of the training instance (as per XGBoost’s scikit-learn API). This is similar to how a random forest operates but without the sample replacement part
+
eta (in XGBoost) or learning_rate (in scikit-learn wrapper interface for XGBoost) - a shrinkage factor applied to alpha (a factor derived from weighted errors) in the boosting algorithm or it simply may be more easily understood as the boosting learning rate used to prevent overfitting
+
+
There are of course a whole bunch of other XGBoost parameters that can be tuned, and in order to keep this post at a reasonable reading length, I won’t go through every single one of them, but see this link as an example parameter set for XGBClassifier().
+
In scikit-learn, there are also two types of gradient boosted tree methods, GradientBoostingClassifer() and HistGradientBoostingClassifier(), in its sklearn.ensemble module (note: equivalent regressor class also available). One way to choose between them is to check sample size first. GradientBoostingClassifer() class is likely better when there is only a small sample size (e.g. when number of sample is less than 10,000), while HistGradientBoostingClassifier() class is likely better when your sample size is at least 10,000+ or more. The HistGradientBoostingClassifier() is a histogram-based gradient boosting classification tree that is mainly inpired by LightGBM.
+
+
+
+
+
A demo
+
In the example below, I’m only using AdaBoost classifier and XGBoost classifier for now. Please note that the dataset used here is very small and the example is likely not going to reflect real-life use case completely (use with care).
xgboost version used: 2.0.3
+scikit-learn version used: 1.5.0
+rdkit version used: 2023.09.6
+
+
+
+
# Show Python version used
+print(sys.version)
+
+
3.11.0 (v3.11.0:deaf509e8f, Oct 24 2022, 14:43:23) [Clang 13.0.0 (clang-1300.0.29.30)]
+
+
+
+
+
+
Data source
+
Data source is based on ChEMBL database version 33 (as shown by the file name below, “chembl_d_ache_33”), which was downloaded previously from last post (on random forest classifier) by using chembl_downloader.
+
+
from pathlib import Path
+
+# Pick any directory, but make sure it's relative to your home directory
+directory = Path.home().joinpath(".data", "blog")
+# Create the directory if it doesn't exist
+directory.mkdir(exist_ok=True, parents=True)
+
+# Create a file path that corresponds to the previously cached ChEMBL data
+path = directory.joinpath(f"chembl_d_ache_33.tsv")
+
+# alternative way to download latest ChEMBL version
+# please see post link - https://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/2_random_forest_classifier.html#data-retrieval-using-chembl_downloader for details
+# note: need to specify late_version = latest() first
+# path = directory.joinpath(f"chembl_d_ache_{latest_version}.tsv")
+
+if path.is_file():
+# If the file already exists, load it
+ df_ache = pd.read_csv(path, sep=',')
+else:
+# If the file doesn't already exist, make the query then cache it
+ df_ache = chembl_downloader.query(sql)
+ df_ache.to_csv(path, sep=",", index=False)
The definition of “NaN” assigned to max_phase indicates that compounds labelled as “NaN” or “null” have no evidence of showing they’ve reached clinical trials yet, but I’m still keeping them in the dataset (this can also be excluded depending on project goals).
+
A max_phase of -1 is assigned to molecules with unknown clinical phase status (ChEMBL reference), for which I’ll drop for this particular experiment.
+
+
# Fill "NaNs" as "0" first
+df_ache.fillna({"max_phase": 0}, inplace=True)
+
+
+
# Select only mols with max_phase of 0 and above
+df_ache = df_ache[(df_ache["max_phase"] >=0)]
# Convert max_phase from float to int for the ease of reading predicted outcomes,
+# otherwise it'll look like "4., 2., 4., ..."
+df_ache = df_ache.astype({"max_phase": int, "canonical_smiles": "string"})
Please note: the only molecule with max_phase of “0.5” was converted into “0” after I’ve converted the datatype of max_phase from float to integer (I’ve left it deliberately like this since this is only a demonstration on using scikit_learn’s pipeline along with scikit_mol, but in reality this should be handled with care i.e. don’t discard it as different max phase values have different meanings!). Therefore the following max_phase value count will have 6411 molecules with max_phase “0”, rather than the previous number of 6410.
Binary classification has been used in my previous posts, e.g. target as max_phase 4 (re-labelled as “1”) with training set of max_phase “null” re-labelled as “0” with their different RDKit molecular features. This time I’ll be using multi-class classification to predict a training set of molecules containing max_phase of 0, 1, 2, 3 and 4.
+
+
+
Define X and y variables
+
+
# A sanity check and view on the original dataset
+df_ache
+
+
+
+
+
+
+
+
+
assay_chembl_id
+
target_type
+
tax_id
+
chembl_id
+
canonical_smiles
+
molecule_chembl_id
+
max_phase
+
standard_type
+
pchembl_value
+
+
+
+
+
0
+
CHEMBL1909212
+
SINGLE PROTEIN
+
9606
+
CHEMBL220
+
CC/C(=C(/CC)c1ccc(O)cc1)c1ccc(O)cc1
+
CHEMBL411
+
4
+
IC50
+
4.59
+
+
+
1
+
CHEMBL1003053
+
SINGLE PROTEIN
+
9606
+
CHEMBL220
+
COc1c2occc2cc2ccc(=O)oc12
+
CHEMBL416
+
4
+
IC50
+
4.27
+
+
+
2
+
CHEMBL2406149
+
SINGLE PROTEIN
+
9606
+
CHEMBL220
+
COc1c2occc2cc2ccc(=O)oc12
+
CHEMBL416
+
4
+
IC50
+
6.12
+
+
+
4
+
CHEMBL3071788
+
SINGLE PROTEIN
+
9606
+
CHEMBL220
+
O=c1cc(-c2ccc(O)cc2)oc2cc(O)cc(O)c12
+
CHEMBL28
+
0
+
IC50
+
7.92
+
+
+
5
+
CHEMBL1119333
+
SINGLE PROTEIN
+
9606
+
CHEMBL220
+
CNCCC(Oc1ccc(C(F)(F)F)cc1)c1ccccc1
+
CHEMBL41
+
4
+
IC50
+
6.89
+
+
+
...
+
...
+
...
+
...
+
...
+
...
+
...
+
...
+
...
+
...
+
+
+
7139
+
CHEMBL5216374
+
SINGLE PROTEIN
+
9606
+
CHEMBL220
+
CCC(C)(C)NCC(O)c1cc(O)cc(OC(=O)N(C)C)c1
+
CHEMBL5220560
+
0
+
IC50
+
4.70
+
+
+
7140
+
CHEMBL5216425
+
SINGLE PROTEIN
+
9606
+
CHEMBL220
+
CC(C)(C)OC(=O)Nc1ccc(O)c(C(=O)NCCCN2CCCCC2)c1
+
CHEMBL5220695
+
0
+
Ki
+
6.92
+
+
+
7141
+
CHEMBL5216408
+
SINGLE PROTEIN
+
9606
+
CHEMBL220
+
O=C1CCOc2cc(OCCCCCSC(=S)N3CCCCC3)ccc21
+
CHEMBL5220742
+
0
+
IC50
+
7.00
+
+
+
7142
+
CHEMBL5218078
+
SINGLE PROTEIN
+
9606
+
CHEMBL220
+
Cn1ccc2cc(-c3cnc4ccc(C(=O)N5CCCCC5)cc4n3)ccc2c1=O
+
CHEMBL5220884
+
0
+
IC50
+
5.27
+
+
+
7143
+
CHEMBL5216374
+
SINGLE PROTEIN
+
9606
+
CHEMBL220
+
COC(=O)c1cc2c(OC(=O)N(C)C)cccc2[n+]([11CH3])c1
+
CHEMBL5220983
+
0
+
IC50
+
7.70
+
+
+
+
7075 rows × 9 columns
+
+
+
+
+
X = df_ache.canonical_smiles
+y = df_ache.max_phase
This post is going to focus on Scikit_mol which has a manual way to handle SMILES errors, as shown in the code below. Another useful way to deal with SMILES errors is Molpipeline’s SMILES error handling, with an example shown in one of its notebooks. The main difference from what I can see, even though I haven’t got to use it yet, is that Molpipeline takes into account of all the invalid SMILES by giving each invalid SMILES a “NaN” label in the pipeline process - this maintains the matrix shape and good for tracking down the problematic SMILES (molecules).
Also, manually checking for any “NaNs” in the canonical SMILES column (X variable), since AdaBoost classifier won’t accept missing values in the dataset, but if using HistGradientBoostingClassifier() instead, it should take care of the native NaNs.
+
+
print(f"{df_ache.canonical_smiles.isna().sum()} out of {len(df_ache)} SMILES failed in conversion")
+
+
0 out of 7075 SMILES failed in conversion
+
+
+
There are other ways to deal with NaNs with a few examples provided by scikit-learn. However, with regards to drug discovery data, there are probably more caveats that need to be taken during data preprocessing (I’m also still exploring this too).
+
+
+
+
Split data
+
Randomly splitting data this time.
+
+
# Found a silly error when naming X, y train/test sets!
+# Remember to name them in this order: X_train, X_test, y_train, y_test
+# otherwise model fitting won't work...
+X_train, X_test, y_train, y_test = train_test_split(X_valid, y, test_size=0.2, random_state=3)
+
+
+
+
+
+
Create pipelines
+
The aim is to create pipeline(s) using scikit-learn.
+
+
+
AdaBoost classifier
+
The original plan is to chain an AdaBoost classifier, XGBoost classifier, along with Scikit-mol transformers all at once. However, it turns out that I’m building two separate pipelines of AdaBoost classifier and XGBoost classifier so that I can compare the difference(s) between them, and this also serves better for the purpose of this post really.
+
This is also the time to think about generating molecular features for model training. Choosing data features such as fingerprints (e.g. Morgan fingerprints which is usually best for larger dataset) or RDKit 2D descriptors (which is useful for smaller datasets) or others. For RDKit 2D descriptors, Scikit_mol has integrated RDKit’s rdkit.Chem.Descriptors module and rdkit.ML.Descriptors.MoleculeDescriptors module within its MolecularDescriptorTransformer().
+
Some useful links regarding building pipelines in scikit-learn and also another reference notebook on when to use parallel calculations for different molecular features:
For the first sample pipeline I’m building below, I’ve noticed that not all of the 209 RDKit 2D descriptors can be used for AdaBoost classifier, as some of the descriptors will have values of “0” and AdaBoost classifier will not be able to take care of them. Therefore, I’m only using a small selection of descriptors only, but HistGradientBoostingClassifier() should be able to take into account of NaNs and can be chained to include all descriptors in a pipeline.
+
The following is an example of building a scikit_learn pipeline by using AdaBoost classifier model, along with Scikit_mol’s transformers for multi-class max_phase predictions with training set consisting of molecules with max_phase 0, 1, 2, 3 and 4. I’ve used Morgan fingerprints instead eventually so that’ll be shown in the following pipeline code, but I’ve also kept the RDKit 2D descriptor option on (just need to uncomment to run).
+
+
# Set parameters for RDKit 2D descriptors
+# params_rdkit2d = {
+# "desc_list": ['HeavyAtomCount', 'FractionCSP3', 'RingCount', 'MolLogP', 'MolWt']
+# }
+
+# Set parameters for adaboost model
+params_adaboost = {
+"estimator": DecisionTreeClassifier(max_depth =3),
+# default: n_estimators = 50, learning_rate = 1.0 (trade-off between them)
+"n_estimators": 80,
+"learning_rate": 0.2,
+# SAMME (Stagewise Additive Modeling using a Multi-class Exponential loss function) algorithm
+# for multi-class classification
+"algorithm": "SAMME",
+"random_state": 2,
+ }
+
+# Building AdaBoostClassifier pipeline
+mlpipe_adaboost = make_pipeline(
+# Convert SMILES to RDKit molecules
+ SmilesToMolTransformer(),
+# Molecule standardisations
+ Standardizer(),
+## A choice of using either Morgan fingerprints or RDKit 2D descriptors:
+# Generate MorganFingerprintTransformer()
+ MorganFingerprintTransformer(useFeatures=True),
+# Generate RDKit2D descriptors
+#MolecularDescriptorTransformer(**params_rdkit2d),
+# Scale variances in descriptor data
+ StandardScaler(),
+# Apply adaptive boost classifier
+ AdaBoostClassifier(**params_adaboost)
+)
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
An interactive pipeline diagram should show with a status of “not fitted” if you hover the mouse over the “i” logo on top right. The pipeline is then fitted onto the training sets.
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
The pipeline status should now show a “fitted” message if hovering over the same “i” logo. Then the pipeline is used on the X_test (testing X set) to predict the target (max_phase) variable.
+
+
mlpipe_adaboost.predict(X_test)
+
+
array([0, 0, 0, ..., 0, 0, 0])
+
+
+
+
+
+
XGBoost classfier
+
The following code snippet is an example of a scikit_learn pipeline using Scikit_mol’s transformers and XGBoost classifier. One nice thing about XGBoost is that is has scikit_learn interface so that it can be integrated into the scikit-learn pipeline and Scikit_mol’s transformers, which is what I’ve tried below.
+
+
# Set parameters for xgboost model
+params_xgboost = {
+"n_estimators": 100,
+"max_depth": 3,
+# For multi-class classification, use softprob for loss function (learning task parameters)
+# source: https://xgboost.readthedocs.io/en/latest/parameter.html#learning-task-parameters
+"objective": 'multi:softprob',
+"learning_rate": 0.1,
+"subsample": 0.5,
+"random_state": 2
+ }
+
+# Building XGBoostClassifier pipeline
+mlpipe_xgb = make_pipeline(
+# Convert SMILES to RDKit molecules
+ SmilesToMolTransformer(),
+# Molecule standardisations
+ Standardizer(),
+## A choice of using either Morgan fingerprints or RDKit 2D descriptors:
+# Generate MorganFingerprintTransformer()
+ MorganFingerprintTransformer(useFeatures=True),
+# Generate RDKit2D descriptors
+#MolecularDescriptorTransformer(**params_rdkit2d),
+# Scale variances in descriptor data
+ StandardScaler(),
+# XGBoost classifier
+ XGBClassifier(**params_xgboost)
+)
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
One can never just leave the process of building a machine learning model without evaluating it. Although what I have done below is probably very minimal but it’s somewhat a starting point to think about how good the model is.
+
+
from sklearn.metrics import accuracy_score
+
+# Following misclassification score function code borrowed and adapted from:
+# https://scikit-learn.org/stable/auto_examples/ensemble/plot_adaboost_multiclass.html#sphx-glr-auto-examples-ensemble-plot-adaboost-multiclass-py
+
+def misclassification_error(y_true, y_pred):
+return1- accuracy_score(y_true, y_pred)
+
+mlpipe_adaboost_misclassification_error = misclassification_error(
+ y_test, mlpipe_adaboost.fit(X_train, y_train).predict(X_test)
+)
+
+mlpipe_xgb_misclassifiaction_error = misclassification_error(
+ y_test, mlpipe_xgb.fit(X_train, y_train).predict(X_test)
+)
+
+print("Training score for mlpipe_adaboost: "f"{mlpipe_adaboost.score(X_train, y_train):0.2f}")
+print("Testing score for mlpipe_adaboost: "f"{mlpipe_adaboost.score(X_test, y_test):0.2f}")
+print("AdaBoostClassifier's misclassification_error: "f"{mlpipe_adaboost_misclassification_error:0.3f}")
+
+print("Training score for mlpipe_xgb: "f"{mlpipe_xgb.score(X_train, y_train):0.2f}")
+print("Testing score for mlpipe_xgb: "f"{mlpipe_xgb.score(X_test, y_test):0.2f}")
+print("XGBClassifier's missclassification_error: "f"{mlpipe_xgb_misclassifiaction_error:0.3f}")
+
+
Training score for mlpipe_adaboost: 0.97
+
+
+
Testing score for mlpipe_adaboost: 0.97
+AdaBoostClassifier's misclassification_error: 0.028
+
+
+
Training score for mlpipe_xgb: 0.99
+
+
+
Testing score for mlpipe_xgb: 0.99
+XGBClassifier's missclassification_error: 0.014
+
+
+
It appears that XGBoost model obtained a better prediction accuracy than the AdaBoost one (although the models are built in a very simple way, but it still shows the slight difference in performance). The training data being used here is also very imbalanced with a lot of them being max_phase of “0” than “4”, and with max_phase “4” being our ultimate aim, the dataset used above is really for demonstration only. Also, since this post is already quite long, I’d rather not make this post into a gigantic tl;dr, so for the imbalanced data discussion and exploration, my previous posts have tried to touch on this topic - “Random forest” and “Random forest classifier”.
+
+
+
+
Hyperparameter tuning for XGBoost classifier
+
For XGBoost, one of the main things is to minimise model overfitting where several parameters will play important roles to achieve this. For example, learning_rate and subsample are the first two mentioned previously, and another technique is based on regularisation which includes two other parameters, reg_alpha (L1 regularisation based on Manhattan distance) and reg_lamda (L2 regularisation based on Euclidean distance). Both of these regularisation parameters aim to penalise XGBoost’s model complexity to make it a bit more conservative in order to reduce overfitting (Bruce, Bruce, and Gedeck 2020).
+
A full list of XGBoost classifier pipeline parameters and settings used can be retrieved as shown below. It contains a long list of parameters and one of the ways to find the optimal set of parameters is by using cross-validation (CV).
To see the default values or types of each XGBoost parameter, this XGBoost documentation link is useful (which can be cross-referenced with XGBoost’s Python API reference).
+
+
+
+
# To obtain only the parameter names for the ease of reading
+mlpipe_xgb.get_params().keys()
Some of the main XGBoost parameters that can be tuned are n_estimators, max_depth, learning_rate, subsample and reg_lambda. Here, I’m going to try to look for the best combination of learning_rate and subsample for a XGBoost classifier model for now.
+
+
# Specify parameters and distributions to be sampled
+params_dist = {
+# learning_rate usually between 0.01 - 0.1 as suggested by Raschka et al.
+# default is between 0 and 1
+"xgbclassifier__learning_rate": [0.05, 0.1, 0.3],
+# subsample default is between 0 and 1
+"xgbclassifier__subsample": [0.5, 0.7, 1.0]
+}
+
+
+
+
+
Randomised search CV
+
The following chunk of code is an example of running randomised search CV. I’ve deliberately folded the code to minimise the reading length of the post and also because the result from it is very similar to the grid search CV used below (randomised search CV run time was 13 min 33.2 seconds due to having two pipelines containing two different machine learning models for the same set of data). It’s being kept as a code reference for anyone who’d like to try it and also as an alternative way to do hyperparameter tuning.
+
+
+Code
+
## Uncomment code below to run
+# from sklearn.model_selection import RandomizedSearchCV
+# from time import time
+
+## Borrowing a utility function code from scikit_learn documentation to report best scores
+## Source: https://scikit-learn.org/stable/auto_examples/model_selection/plot_randomized_search.html#sphx-glr-auto-examples-model-selection-plot-randomized-search-py
+
+# def report(results, n_top=3):
+# for i in range(1, n_top + 1):
+# candidates = np.flatnonzero(results["rank_test_score"] == i)
+# for candidate in candidates:
+# print("Model with rank: {0}".format(i))
+# print(
+# "Mean validation score: {0:.3f} (std: {1:.3f})".format(
+# results["mean_test_score"][candidate],
+# results["std_test_score"][candidate],
+# )
+# )
+# print("Parameters: {0}".format(results["params"][candidate]))
+# print("")
+
+## The following code has also referenced and adapted from this notebook
+## https://github.com/EBjerrum/scikit-mol/blob/main/notebooks/06_hyperparameter_tuning.ipynb
+
+# n_iter_search = 9
+
+# random_search = RandomizedSearchCV(
+# mlpipe_xgb,
+# param_distributions=params_dist,
+# n_iter=n_iter_search,
+# n_jobs=2
+# )
+
+# t_start = time()
+# random_search.fit(X_train, y_train)
+# t_finish = time()
+
+# print(f'Runtime: {t_finish-t_start:0.2F} seconds for {n_iter_search} iterations')
+
+## Run report function code
+# report(random_search.cv_results_)
+
+
+
+
+
+
Grid search CV
+
+
grid_search = GridSearchCV(
+ mlpipe_xgb,
+ param_grid=params_dist,
+ verbose=1,
+ n_jobs=2
+)
+
+grid_search.fit(X_train, y_train)
+
+print(f"The best cv score is: {grid_search.best_score_:0.2f}")
+print(f"The best cv parameter settings are: {grid_search.best_params_}")
+
+# This may take longer time to run depending on computer hardware specs (for me it's taken ~13min)
+
+
Fitting 5 folds for each of 9 candidates, totalling 45 fits
+
+
+
The best cv score is: 0.99
+The best cv parameter settings are: {'xgbclassifier__learning_rate': 0.3, 'xgbclassifier__subsample': 0.7}
+
+
+
For tuning parameters of Morgan fingerprints, this Scikit_mol example notebook explains how to do it with code, so I won’t repeat them here, but have only shown how to tune some of the main XGBoost parameters.
+
+
+
+
Pickle model
+
The next step is to pickle the model or pipeline if wanting to save it for future use and to avoid re-training model from the ground up again.
One thing to remember is to avoid unpickling unknown files over insecure network, and add security key if needed.
+
+
# Pickle to save (serialise) the model in working directory (specify path if needed)
+# "wb" - write binary
+pickle.dump(mlpipe_xgb, open("xgb_pipeline.pkl", "wb"))
+# Unpickle (de-serialise) the model
+# "rb" - read binary
+mlpipe_xgb_2 = pickle.load(open("xgb_pipeline.pkl", "rb"))
+
+# Use the unpickled model object to make prediction
+pred2 = mlpipe_xgb_2.predict(X_test)
+
+## Check unpickled model and original model are the same via Python's assertion method
+#assert np.sum(np.abs(pred2 - pred)) == 0
+## or alternatively use numpy's allclose()
+print(np.allclose(pred, pred2))
+
+
True
+
+
+
+
+
+
+
Acknowledgement
+
Again, this grows into another really long post… Although this post has taken quite a long time to build up to completion, I still want to thank all the contributors or developers for all the packages used in this post.
+Chen, Tianqi, and Carlos Guestrin. 2016. “XGBoost: A Scalable Tree Boosting System.”CoRR abs/1603.02754. http://arxiv.org/abs/1603.02754.
+
+
+Freund, Yoav, and Robert E Schapire. 1997. “A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting.”Journal of Computer and System Sciences 55 (1): 119–39. https://doi.org/10.1006/jcss.1997.1504.
+
+
+Raschka, Sebastian, Yuxi (Hayden) Liu, and Vahid Mirjalili. 2022. Machine Learning with PyTorch and Scikit-Learn. Birmingham, UK: Packt Publishing.
+
]]>
+ Machine learning projects
+ Tree models
+ Pandas
+ Scikit-learn
+ RDKit
+ ChEMBL database
+ Python
+ https://jhylin.github.io/Data_in_life_blog/posts/19_ML2-3_Boosted_trees/1_adaboost_xgb.html
+ Wed, 05 Jun 2024 12:00:00 GMT
+
+
+ Using Molstar in Quarto
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/18_Notes_molstar_quarto/Molstar_quarto.html
+
+
Background
+
This is really a short post (note) for myself and probably for others who may be interested in software tools to visualise in silico macromolecules and small molecules.
+
Most bioinformaticians or structural biologists are probably already familiar with this software package, Molstar or Mol* (Sehnal et al. 2021). Molstar is a 3D viewer for large macromolecules (e.g. proteins), which are commonly used in structural biology and drug discovery (and also other related scientific disciplines).
+
A Quarto extension has been developed to embed the Molstar interactive 3D viewer inside Quarto markdown documents, which can be rendered as HTML pages. The main advantage of this is that it’s useful for reports or presentations.
Streamlit and Dash integrations are also possible, this also makes me think that I could probably try integrating Molstar with Shiny for Python, it’ll likely be a future side project.
+
+
+
+
+
An example using Molstar with RCSB PDB
+
The following example retrieves a protein (PDB ID: 4MQT) from RCSB PDB.
+
{{< mol-rcsb 4mqt >}}
+
+
+
+
Hover over protein structure to see details of amino acid residues or ligands present in the structure.
+
To focus or zoom-in on the ligand bound to the receptor, just click on the ligand first. This shows most of the chemical interactions between the receptor and ligand bound to it (e.g. hydrogen bondings, other chemical interactions will appear if present e.g. pi-pi stacking).
+
Screenshots or state snapshots are also available from the viewer (other utility functions can be found on the top right corner of the viewer).
+
+
+
+An close-up screenshot of 4MQT showing bound ligand
+
+
+
MD trajectories are also available, although I haven’t quite got there yet but it’s useful to know this may be possible (see example C from https://molstar.org/viewer-docs/examples/).
+
It’s also possible to upload AlphaFold-sourced proteins, or from other file sources (see examples shown from Molstar example).
+
+
+
+
+
+
References
+
+Sehnal, David, Sebastian Bittrich, Mandar Deshpande, Radka Svobodová, Karel Berka, Václav Bazgier, Sameer Velankar, Stephen K Burley, Jaroslav Koča, and Alexander S Rose. 2021. “Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures.”Nucleic Acids Research 49 (W1): W431–37. https://doi.org/10.1093/nar/gkab314.
+
+
]]>
+ Notes
+ Molecular viz
+ https://jhylin.github.io/Data_in_life_blog/posts/18_Notes_molstar_quarto/Molstar_quarto.html
+ Fri, 05 Apr 2024 11:00:00 GMT
+
+
+
+ Random forest classifier
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/2_random_forest_classifier.html
+ The section on “Data retrieval using chembl_downloader” has been updated and finalised on 31st January 2024 - many thanks for the comment from Charles Tapley Hoyt (cthoyt).
+
This post was really just an addition towards the last random forest (RF) post. It was mainly inspired by this paper (Esposito et al. 2021) from rinikerlab1. It was nice to complete the RF series by adding a RF classifier since last post was only on a regressor. Another thing was that imbalanced datasets were common in drug discovery projects, learning different strategies to deal with them was also very useful. While working on this post, I also came across a few other packages that I haven’t used before so I’ve included them all down below.
+
+
+
+
Overview of post
+
+
Data sourcing via chembl_downloader
+
Minor data preprocessing using own little script and also SMILES checker from scikit_mol
+
scikit-learn’s RandomForestClassifier()
+
Dealing with imbalanced dataset in RF classifiers by using ghostml
+
A small section on plotting receiver operating characteristic (ROC) curves
chembl_downloader was something I wanted to try a while back. I’ve tried manual download and chembl_webresource_client previously, and they were probably not the best strategies for data reproducibility. The idea of chembl_downloader was to generate a reproducible ChEMBL data source. It involved some SQL at the beginning to specify the exact type of data needed, so some SQL knowledge were required. Other uses for this package were elaborated much more clearly in its GitHub repository at https://github.com/cthoyt/chembl-downloader. One of the reference notebooks that I’ve used could be reached here (more available in its repository).
+
What I did was shown below.
+
+
# Show the latest version of ChEMBL used
+latest_version = latest()
+print(f"The latest ChEMBL version is: {latest_version}")
+
+
The latest ChEMBL version is: 33
+
+
+
The following section was updated as suggested by cthoyt (via his comment for post below). I ended up putting through my first ever pull request in an open-source and cheminformatics-related repository. A new option to choose max_phase was added into the get_target_sql function in chembl_downloader by keeping it as a togglable option via boolean flag. Many thanks for the patience from cthoyt for guiding me through it. The overall code was now changed as shown below.
+
::: {.cell execution_count=3}
+
# Generate SQL for a query on acetylcholinesterase (AChE): CHEMBL220
+sql = queries.get_target_sql(target_id="CHEMBL220", target_type="SINGLE PROTEIN", max_phase=True)
+
+# Pretty-print the SQL in Jupyter
+queries.markdown(sql)
+
+
SELECT
+ ASSAYS.chembl_id AS assay_chembl_id,
+ TARGET_DICTIONARY.target_type,
+ TARGET_DICTIONARY.tax_id,
+ COMPOUND_STRUCTURES.canonical_smiles,
+ MOLECULE_DICTIONARY.chembl_id AS molecule_chembl_id,
+ MOLECULE_DICTIONARY.max_phase,
+ ACTIVITIES.standard_type,
+ ACTIVITIES.pchembl_value
+FROM TARGET_DICTIONARY
+JOIN ASSAYS ON TARGET_DICTIONARY.tid == ASSAYS.tid
+JOIN ACTIVITIES ON ASSAYS.assay_id == ACTIVITIES.assay_id
+JOIN MOLECULE_DICTIONARY ON MOLECULE_DICTIONARY.molregno == ACTIVITIES.molregno
+JOIN COMPOUND_STRUCTURES ON MOLECULE_DICTIONARY.molregno == COMPOUND_STRUCTURES.molregno
+WHERE TARGET_DICTIONARY.chembl_id ='CHEMBL220'
+AND ACTIVITIES.pchembl_value ISNOTNULL
+AND TARGET_DICTIONARY.target_type ='SINGLE PROTEIN'```
+:::
+:::
+
+
+::: {.cell execution_count=4}
+``` {.python .cell-code}
+# The SQL still works as shown above,
+# please ignore the non-SQL texts
+# - bit of a formatting issue
+# when pretty-printing the sql,
+# but shouldn't affect the code
+
+
I’ve also updated how I retrieved and saved the ChEMBL data with the following code suggested and provided by cthoyt. This would be a better and more reproducible way for anyone who might be interested in re-running this notebook.
+
+
from pathlib import Path
+
+# Pick any directory, but make sure it's relative to your home directory
+directory = Path.home().joinpath(".data", "blog")
+# Create the directory if it doesn't exist
+directory.mkdir(exist_ok=True, parents=True)
+
+# Create a file path that corresponds to the version, since this could change
+path = directory.joinpath(f"chembl_d_ache_{latest_version}.tsv")
+
+if path.is_file():
+# If the file already exists, load it
+ df_ache = pd.read_csv(path, sep=',')
+else:
+# If the file doesn't already exist, make the query then cache it
+ df_ache = chembl_downloader.query(sql)
+ df_ache.to_csv(path, sep=",", index=False)
+
+
The rest of the code outputs in the post stayed the same as before. The only thing changed and updated was the part on retrieving ChEMBL data via chembl_downloader.
+
+
+
+
Some data cleaning
+
Minor cleaning and preprocessing were done for this post only, as the focus was more on dealing with imbalanced dataset in RF classifier. Since I used a different way to retrieve ChEMBL data this time, the dataset used here might be slightly different from the one used in previous post.
+
+
+
mol_prep.py
+
I’ve more or less accumulated small pieces of code over time, and I’ve decided to compile them into a Python script. The idea was to remove most function code in the post to avoid repeating them all the time since they’ve been used frequently in the last few posts. The script would be saved into the RF repository, and would still be considered as a “work-in-progress” script (needs more work in the future).
+
+
## Trial own mol_prep.py script
+from mol_prep import preprocess, rdkit_2d_descriptors
scikit_mol was a package originated from RDKit UGM hackathon in 2022. This blog post elaborated further on its functions and uses in machine learning. For this post I’ve only used it for a very small portion, mainly to check for missing SMILES or errors in SMILES (kind of like double checking whether the preprocess function code worked as expected). It could be integrated with scikit-learn’s pipeline method for multiple estimators. Its GitHub Repository link: https://github.com/EBjerrum/scikit-mol - I’ve referred to this reference notebook while working on this post.
+
+
# Quick simple way to check for missing SMILES
+print(f'Dataset contains {df_prep.standard_smiles.isna().sum()} unparsable mols')
+
+
Dataset contains 0 unparsable mols
+
+
+
+
# Checking for invalid SMILES using scikit_mol
+smileschecker = CheckSmilesSanitazion()
+smileschecker.sanitize(list(df_prep.standard_smiles))
+
+# Showing SMILES errors
+smileschecker.errors
+
+
+
+
+
+
+
+
+
SMILES
+
+
+
+
+
+
+
+
+
It showed no errors in SMILES (errors should be listed in the code cell output).
A different spreads of max phases were shown this time in the dataframe, as the SQL query mainly used IC50, whereas last post was strictly limited to Ki via ChEMBL web resource client. Other likely reason was that in the decision tree series, I’ve attempted data preprocessing at a larger scale so some data were eliminated. It appeared that there were more max phase 4 compounds here than last time (note: null compounds were not shown in the value counts as it was labelled as “NaN”, it should be the largest max phase portion in the data).
+
+
# Find out counts of each max phase
+df_merge.value_counts("max_phase")
# Dropping duplicated compound via chembl IDs in the main df
+df_merge_new = df_merge.drop_duplicates(subset=["molecule_chembl_id"], keep="first")
+print(df_merge_new.shape)
+df_merge_new.head()
+
+
(5357, 212)
+
+
+
+
+
+
+
+
+
+
max_phase
+
molecule_chembl_id
+
MaxAbsEStateIndex
+
MaxEStateIndex
+
MinAbsEStateIndex
+
MinEStateIndex
+
qed
+
SPS
+
MolWt
+
HeavyAtomMolWt
+
...
+
fr_sulfide
+
fr_sulfonamd
+
fr_sulfone
+
fr_term_acetylene
+
fr_tetrazole
+
fr_thiazole
+
fr_thiocyan
+
fr_thiophene
+
fr_unbrch_alkane
+
fr_urea
+
+
+
+
+
0
+
4.0
+
CHEMBL411
+
9.410680
+
9.410680
+
0.284153
+
0.284153
+
0.779698
+
12.100000
+
268.356
+
248.196
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
1
+
4.0
+
CHEMBL416
+
11.173100
+
11.173100
+
0.405828
+
-0.405828
+
0.586359
+
11.062500
+
216.192
+
208.128
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
3
+
-1.0
+
CHEMBL7002
+
11.591481
+
11.591481
+
0.189306
+
-0.309798
+
0.886859
+
23.608696
+
333.453
+
310.269
+
...
+
1
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
4
+
NaN
+
CHEMBL28
+
12.020910
+
12.020910
+
0.018823
+
-0.410347
+
0.631833
+
10.800000
+
270.240
+
260.160
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
5
+
4.0
+
CHEMBL41
+
12.564531
+
12.564531
+
0.203346
+
-4.329869
+
0.851796
+
12.909091
+
309.331
+
291.187
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
+
5 rows × 212 columns
+
+
+
+
+
# Making sure previously used 10 max phase 4 compounds could be found in df_merge_new
+df_mp4 = df_merge_new.loc[df_merge_new["molecule_chembl_id"].isin(list_mp4)]
+df_mp4
+
+
+
+
+
+
+
+
+
max_phase
+
molecule_chembl_id
+
MaxAbsEStateIndex
+
MaxEStateIndex
+
MinAbsEStateIndex
+
MinEStateIndex
+
qed
+
SPS
+
MolWt
+
HeavyAtomMolWt
+
...
+
fr_sulfide
+
fr_sulfonamd
+
fr_sulfone
+
fr_term_acetylene
+
fr_tetrazole
+
fr_thiazole
+
fr_thiocyan
+
fr_thiophene
+
fr_unbrch_alkane
+
fr_urea
+
+
+
+
+
9
+
4.0
+
CHEMBL481
+
13.581173
+
13.581173
+
0.095133
+
-1.863974
+
0.355956
+
22.209302
+
586.689
+
548.385
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
67
+
4.0
+
CHEMBL95
+
6.199769
+
6.199769
+
0.953981
+
0.953981
+
0.706488
+
15.200000
+
198.269
+
184.157
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
249
+
4.0
+
CHEMBL502
+
12.936933
+
12.936933
+
0.108783
+
0.108783
+
0.747461
+
20.214286
+
379.500
+
350.268
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
508
+
4.0
+
CHEMBL640
+
11.743677
+
11.743677
+
0.044300
+
-0.044300
+
0.731540
+
10.529412
+
235.331
+
214.163
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
512
+
4.0
+
CHEMBL659
+
9.972866
+
9.972866
+
0.008380
+
-0.411699
+
0.800524
+
33.857143
+
287.359
+
266.191
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
1013
+
4.0
+
CHEMBL1025
+
12.703056
+
12.703056
+
0.426312
+
-4.304784
+
0.629869
+
13.000000
+
184.147
+
170.035
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
1345
+
4.0
+
CHEMBL1128
+
9.261910
+
9.261910
+
0.000000
+
0.000000
+
0.608112
+
10.692308
+
201.697
+
185.569
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
2028
+
4.0
+
CHEMBL360055
+
6.476818
+
6.476818
+
0.656759
+
0.656759
+
0.205822
+
12.583333
+
510.828
+
450.348
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
2725
+
4.0
+
CHEMBL1677
+
6.199769
+
6.199769
+
0.000000
+
0.000000
+
0.760853
+
14.250000
+
234.730
+
219.610
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
3271
+
4.0
+
CHEMBL1200970
+
2.520809
+
2.520809
+
0.000000
+
0.000000
+
0.709785
+
14.000000
+
348.943
+
323.743
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
+
10 rows × 212 columns
+
+
+
+
+
# note: compounds with max phase 0 not shown in the count
+df_merge_new.value_counts("max_phase")
The aim of this post was to model and classify the max phases of ChEMBL small molecules, i.e. whether the compounds in the testing set (consisted of max phase 0 or null compounds) might be eventually classified as max phase 4 or not. This was one of the approaches to answer the question in mind, and not the ultimate way to solve the problem (just thought to mention). The target was “max_phase” and features to be used were the various RDKit 2D descriptors (RDKit2D).
+
The steps I’ve taken to build the model were shown below:
+
+
Re-labelled max phases as binary labels (e.g. max phase null as 0, max phase 4 as 1)
+
+
+
# Re-label max phase NaN as 0
+df_merge_new = df_merge_new.fillna(0)
+df_merge_new.head()
+
+
+
+
+
+
+
+
+
max_phase
+
molecule_chembl_id
+
MaxAbsEStateIndex
+
MaxEStateIndex
+
MinAbsEStateIndex
+
MinEStateIndex
+
qed
+
SPS
+
MolWt
+
HeavyAtomMolWt
+
...
+
fr_sulfide
+
fr_sulfonamd
+
fr_sulfone
+
fr_term_acetylene
+
fr_tetrazole
+
fr_thiazole
+
fr_thiocyan
+
fr_thiophene
+
fr_unbrch_alkane
+
fr_urea
+
+
+
+
+
0
+
4.0
+
CHEMBL411
+
9.410680
+
9.410680
+
0.284153
+
0.284153
+
0.779698
+
12.100000
+
268.356
+
248.196
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
1
+
4.0
+
CHEMBL416
+
11.173100
+
11.173100
+
0.405828
+
-0.405828
+
0.586359
+
11.062500
+
216.192
+
208.128
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
3
+
-1.0
+
CHEMBL7002
+
11.591481
+
11.591481
+
0.189306
+
-0.309798
+
0.886859
+
23.608696
+
333.453
+
310.269
+
...
+
1
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
4
+
0.0
+
CHEMBL28
+
12.020910
+
12.020910
+
0.018823
+
-0.410347
+
0.631833
+
10.800000
+
270.240
+
260.160
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
5
+
4.0
+
CHEMBL41
+
12.564531
+
12.564531
+
0.203346
+
-4.329869
+
0.851796
+
12.909091
+
309.331
+
291.187
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
+
5 rows × 212 columns
+
+
+
+
+
Splitted data into max phase null & max phase 4 (needing to re-label max phase 4 column only as 1, and not disrupting the labels of max phase 0 compounds)
+
+
+
# Select all max phase null compounds
+df_null = df_merge_new[df_merge_new["max_phase"] ==0]
+print(df_null.shape)
+df_null.head()
+
+
(5256, 212)
+
+
+
+
+
+
+
+
+
+
max_phase
+
molecule_chembl_id
+
MaxAbsEStateIndex
+
MaxEStateIndex
+
MinAbsEStateIndex
+
MinEStateIndex
+
qed
+
SPS
+
MolWt
+
HeavyAtomMolWt
+
...
+
fr_sulfide
+
fr_sulfonamd
+
fr_sulfone
+
fr_term_acetylene
+
fr_tetrazole
+
fr_thiazole
+
fr_thiocyan
+
fr_thiophene
+
fr_unbrch_alkane
+
fr_urea
+
+
+
+
+
4
+
0.0
+
CHEMBL28
+
12.020910
+
12.020910
+
0.018823
+
-0.410347
+
0.631833
+
10.800000
+
270.240
+
260.160
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
8
+
0.0
+
CHEMBL8320
+
10.282778
+
10.282778
+
0.120741
+
-0.120741
+
0.416681
+
17.500000
+
108.096
+
104.064
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
14
+
0.0
+
CHEMBL11833
+
11.201531
+
11.201531
+
0.428520
+
-0.466092
+
0.838024
+
25.157895
+
262.309
+
244.165
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
392
+
0.0
+
CHEMBL12324
+
11.257704
+
11.257704
+
0.462395
+
-0.462395
+
0.797990
+
26.150000
+
277.344
+
256.176
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
393
+
0.0
+
CHEMBL274107
+
11.359778
+
11.359778
+
0.372211
+
-0.473241
+
0.838024
+
25.157895
+
262.309
+
244.165
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
+
5 rows × 212 columns
+
+
+
+
+
# Using pd.DataFrame.assign to add a new column to re-label max_phase 4 as "1"
+df_mp4_lb = df_mp4.assign(max_phase_lb = df_mp4["max_phase"] /4)
+
+# Using pd.DataFrame.pop() & insert() to shift added column to first column position
+first_col = df_mp4_lb.pop("max_phase_lb")
+df_mp4_lb.insert(0, "max_phase_lb", first_col)
+df_mp4_lb.head()
+
+
+
+
+
+
+
+
+
max_phase_lb
+
max_phase
+
molecule_chembl_id
+
MaxAbsEStateIndex
+
MaxEStateIndex
+
MinAbsEStateIndex
+
MinEStateIndex
+
qed
+
SPS
+
MolWt
+
...
+
fr_sulfide
+
fr_sulfonamd
+
fr_sulfone
+
fr_term_acetylene
+
fr_tetrazole
+
fr_thiazole
+
fr_thiocyan
+
fr_thiophene
+
fr_unbrch_alkane
+
fr_urea
+
+
+
+
+
9
+
1.0
+
4.0
+
CHEMBL481
+
13.581173
+
13.581173
+
0.095133
+
-1.863974
+
0.355956
+
22.209302
+
586.689
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
67
+
1.0
+
4.0
+
CHEMBL95
+
6.199769
+
6.199769
+
0.953981
+
0.953981
+
0.706488
+
15.200000
+
198.269
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
249
+
1.0
+
4.0
+
CHEMBL502
+
12.936933
+
12.936933
+
0.108783
+
0.108783
+
0.747461
+
20.214286
+
379.500
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
508
+
1.0
+
4.0
+
CHEMBL640
+
11.743677
+
11.743677
+
0.044300
+
-0.044300
+
0.731540
+
10.529412
+
235.331
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
512
+
1.0
+
4.0
+
CHEMBL659
+
9.972866
+
9.972866
+
0.008380
+
-0.411699
+
0.800524
+
33.857143
+
287.359
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
+
5 rows × 213 columns
+
+
+
+
+
# Also create a new column max_phase_lb column for df_null
+# in order to merge 2 dfs later
+df_null_lb = df_null.assign(max_phase_lb = df_null["max_phase"])
+first_col_null = df_null_lb.pop("max_phase_lb")
+df_null_lb.insert(0, "max_phase_lb", first_col_null)
+df_null_lb.head()
# Convert both X & y to arrays
+X = X.to_numpy()
+y = y.to_numpy()
+
+
+
# Using train_test_split() this time to split data
+X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, stratify=y, random_state=1)
+
+
After data splitting, a RF classifier was trained with reference to this notebook.
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Extracted positive prediction probabilities for the testing set and showed confusion matrix with classification metrics
+
+
+
test_probs = rfc.predict_proba(X_test)[:, 1]
+
+
Some reference links and explanations for area under the ROC curve and Cohen’s Kappa.
+
Area under the ROC curve: reference - the area under a curve plot between sensitivity or recall (percent of all 1s classified correctly by a classifier or true positive rate) and specificity (percent of all 0s classified correctly by a classifier, or equivalent to 1 - false positive rate or true negative rate) (Bruce, Bruce, and Gedeck 2020). It is useful for evaluating the performance of a classification model via comparing the true positive rate and false positive rate which are influenced by shifting the decision threshold. Area under the ROC is usually represented as a number ranging from 0 to 1 (1 being a perfect classifier, 0.5 or below meaning a poor, ineffective classifier)
+
Cohen’s Kappa score: reference - a score that is used to measure the agreement of labelling between two annotators (usually between -1 and 1, the higher the score the better the agreement)
+
Rather than re-inventing the wheel, the following function code for calculating metrics of the RF model were adapted from this notebook, from GHOST repository. I have only added some comments for clarities, and also added a zero_division parameter for the classification_report to mute the warning message when the results ended up being 0 due to divisions by zero.
+
+
def calc_metrics(y_test, test_probs, threshold =0.5):
+# Target label assigned according to stated decision threshold (default = 0.5)
+# e.g. second annotator (expected label)
+ scores = [1if x>=threshold else0for x in test_probs]
+# Calculate area under the ROC curve based on prediction score
+ auc = metrics.roc_auc_score(y_test, test_probs)
+# Calculate Cohen's Kappa score
+# e.g. y_test as first annotator (predicted label)
+ kappa = metrics.cohen_kappa_score(y_test, scores)
+# Formulate the confusion matrix
+ confusion = metrics.confusion_matrix(y_test, scores, labels =list(set(y_test)))
+print('thresh: %.2f, kappa: %.3f, AUC test-set: %.3f'%(threshold, kappa, auc))
+print(confusion)
+print(metrics.classification_report(y_test, scores, zero_division=0.0))
+return
+
+
Note: roc_auc_score measures true positive and false positive rates, requiring binary labels (e.g. 0s and 1s) in the data
+
Then showed confusion matrix along with area under the ROC curve and Cohen’s Kappa.
It was very obvious that not all of the compounds were classified in the testing set. There were only 1052 compounds classified as true negative, and none in the testing set were labelled as true positive. The likely reason was due to the very imbalanced ratio of actives (only 10 max phase 4 which were labelled as “1” compounds) and inactives (5256 max phase 0 compounds). Besides the imbalanced dataset, the decision threshold was also normally set at 0.5, meaning the classifier was likely going to lose the chance to classify the true positive compounds due to the very skewed ratio of actives to inactives.
+
+
Two approaches were used in the GHOST (generalized threshold shifting) paper:
Approach 2 led to GHOST procedure with a goal to optimise and shift the decision threshold in any classification methods to catch the minor portion of actives (rather than the major portion of inactives)
+
note: both approaches were shown to be performing similarly in the paper
+
+
+
I only used approach 2 here since the RDKit blog post had already explained approach 1 in depth.
+
The next step involved extracting prediction probabilities from the RF classifier trained model.
+
+
# Get the positive prediction probabilities of the training set
+train_probs = rfc.predict_proba(X_train)[:, 1]
+
+
+
Used GHOST strategy in a postprocessing way (note: last post used data re-sampling method in a preprocessing way)
+
+
The decision threshold were optimised by using ghostml code via testing various different thresholds, e.g. in spaces of 0.05 that ranged from 0.05 to 0.5. The most optimal threshold would have the most maximised Cohen’s kappa.
+
+
# Setting up different decision thresholds
+thresholds = np.round(np.arange(0.05,0.55,0.05), 2)
+thresholds
# Looking for the best threshold with the most optimal Cohen's Kappa
+new_threshold = ghostml.optimize_threshold_from_predictions(y_train, train_probs, thresholds, ThOpt_metrics ='ROC')
+
+
Using the calc_metrics function again on the newly-found or shifted decision threshold.
Here, after shifting the decision threshold with the most optimal Cohen’s Kappa score, we could see an improved number of compounds labelled within the true negative class (increasing from 1052 to 4204), and more importantly, we could see the true positive class improved from 0 to 7 as well.
+
+
+
+
Plotting ROC curves
+
Time for some plots - I’ve shown two different ways to plot ROC curves below.
+
+
Using scikit-learn
+
+
Testing set ROC curve - obviously, this was not a good classifier with a poor AUC.
I wanted to mention that the testing set used here was most likely not the best ones to be used. There could be many overlaps or similarities between the training and testing sets, since they all came from ChEMBL database. For demonstration and learning purposes, I ended up using similar dataset as last time. Hopefully, I can try other open-source or public drug discovery datasets in the near future.
+
The other thing to mention was that I should try different molecular fingerprints or descriptors as well, rather than only using RDKit2D, which might lead to different results. I should also probably slowly move onto using multiple datasets or targets in a project, which would likely make things more interesting. On the other hand, I also wanted to avoid this in order to make the topic of interest as clear and simple as possible for me or anyone who’s trying to learn.
+
+
+
+
Acknowledgements
+
I’d like to thank Riniker lab again for the GHOST paper, along with all the authors, contributors or developers for all of the software packages mentioned in this post, and also, huge thanks should also go to the authors of the reference notebooks mentioned in the post as well.
+Esposito, Carmen, Gregory A. Landrum, Nadine Schneider, Nikolaus Stiefl, and Sereina Riniker. 2021. “GHOST: Adjusting the Decision Threshold to Handle Imbalanced Data in Machine Learning.”Journal of Chemical Information and Modeling 61 (6): 2623–40. https://doi.org/10.1021/acs.jcim.1c00160.
+
+
Footnotes
+
+
+
h/t: Greg Landrum for his comment on Mastodon for the last RF post (which led to this follow-up post)↩︎
+
+
]]>
+ Machine learning projects
+ Tree models
+ Pandas
+ Scikit-learn
+ RDKit
+ ChEMBL database
+ Python
+ https://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/2_random_forest_classifier.html
+ Tue, 16 Jan 2024 11:00:00 GMT
+
+
+ Random forest
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/1_random_forest.html
+ Post updated on 3rd May 2024 - Added comment regarding ImbalancedLearningRegression package (installation tip) & Jupyter notebook link of this post
+
+
Quick overview of this post
+
+
Short introduction of random forest
+
Random forest methods or classes in scikit-learn
+
Random forest regressor model in scikit-learn
+
Training and testing data splits
+
+
ChEMBL-assigned max phase splits
+
Imbalanced learning regression and max phase splits
+
+
Scoring metrics of trained models
+
Feature importances in dataset
+
+
feature_importances_attribute in scikit-learn
+
permutation_importance function in scikit-learn
+
SHAP approach
+
+
Hyperparameter tuning on number of trees
+
+
+
+
+
What is a random forest?
+
The decision tree model built last time was purely based on one model on its own, which often might not be as accurate or reflective in real-life. To improve the model, the average outcome from multiple models (Breiman 1998) should be considered to see if this would provide a more realistic image. This model averaging approach was also constantly used in our daily lives, for example, using majority votes during decision-making steps.
+
The same model averaging concept was also used in random forest (Breiman 2001), which as the name suggested, was composed of many decision trees (models) forming a forest. Each tree model would be making its own model prediction. By accruing multiple predictions since we have multiple trees, the average obtained from these predictions would produce one single result in the end. The advantage of this was that it improved the accuracy of the prediction by reducing variances, and also minimised the problem of overfitting the model if it was purely based on one model only (more details in section 1.11.2.1. Random Forests from scikit-learn).
+
The “random” part of the random forest was introduced in two ways. The first one was via using bootstrap samples, which was also known as bagging or bootstrap aggregating (Bruce, Bruce, and Gedeck 2020), where samples were drawn with replacements within the training datasets for each tree built in the ensemble (also known as the perturb-and-combine technique (Breiman 1998)). While bootstrap sampling was happening, randomness was also incorporated into the training sets at the same time. The second way randomness was introduced was by using a random subset of features for splitting at the nodes, or a full set of features could also be used (although this was generally not recommended). The main goal here was to achieve best splits at each node.
+
+
+
+
Random forest in scikit-learn
+
Scikit-learn had two main types of random forest classes - ensemble.RandomForestClassifier() and ensemble.RandomForestRegressor(). When to use which class would depend on the target values. The easiest thing to do was to decide whether the target variables had class labels (binary types or non-continuous variables e.g. yes or no, or other different categories to be assigned) or continuous (numerical) variables, which in this case, if I were to continue using the same dataset from the decision tree series, it would be a continuous variable or feature, pKi, the inhibition constant.
+
There were also two other alternative random forest methods in scikit-learn, which were ensemble.RandomTreesEmbedding() and ensemble.ExtraTreesClassifier() or ensemble.ExtraTreesRegressor(). The difference for RandomTreesEmbedding() was that it was an unsupervised method that used data transformations (more details from section 1.11.2.6. on “Totally Random Trees Embedding” in scikit-learn). On the other side, there was also an option to use ExtraTreesClassifier() or ExtraTreesRegressor() to generate extremely randomised trees that would go for another level up in randomness (more deatils in section 1.11.2.2. on Extremely Randomized Trees from scikit-learn). The main difference for this type of random forest was that while there was already a random subset of feature selection used (with an intention to select the most discerning features), more randomness were added on top of this by using purely randomly generated splitting rules for picking features at the nodes. The advantage of this type of method was that it would reduce variance and increase the accuracy of the model, but the downside was there might be an increase in bias within the model.
+
+
+
+
Building a random forest regressor model using scikit-learn
+
As usual, all the required libraries were imported first.
+
+
import pandas as pd
+import sklearn
+from sklearn.ensemble import RandomForestRegressor
+
+# For imbalanced datasets in regression
+# May need to set env variable (SKLEARN_ALLOW_DEPRECATED_SKLEARN_PACKAGE_INSTALL=True) when installing
+# due to package dependency on older sklearn version
+import ImbalancedLearningRegression as iblr
+
+# Plots
+import matplotlib.pyplot as plt
+import seaborn as sns
+
+# Metrics
+from sklearn.metrics import mean_squared_error
+from sklearn.metrics import r2_score
+
+# Feature importances
+# Permutation_importance
+from sklearn.inspection import permutation_importance
+# SHAP values
+import shap
+
+# Hyperparameter tuning
+from sklearn.model_selection import cross_val_score, RepeatedKFold
+
+from numpy import mean, std
+from natsort import index_natsorted
+import numpy as np
+
+# Showing version of scikit-learn used
+print(sklearn.__version__)
+
+
1.3.2
+
+
+
Importing dataset that was preprocessed from last time - link to data source: first decision tree post.
+
+
data = pd.read_csv("ache_2d_chembl.csv")
+data.drop(columns = ["Unnamed: 0"], inplace=True)
+# Preparing data for compounds with max phase with "NaN" by re-labelling to "null"
+data["max_phase"].fillna("null", inplace=True)
+data.head()
+
+
Setting an item of incompatible dtype is deprecated and will raise in a future error of pandas. Value 'null' has dtype incompatible with float64, please explicitly cast to a compatible dtype first.
+
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
pKi
+
max_phase
+
mw
+
fsp3
+
n_lipinski_hba
+
n_lipinski_hbd
+
n_rings
+
n_hetero_atoms
+
n_heavy_atoms
+
...
+
sas
+
n_aliphatic_carbocycles
+
n_aliphatic_heterocyles
+
n_aliphatic_rings
+
n_aromatic_carbocycles
+
n_aromatic_heterocyles
+
n_aromatic_rings
+
n_saturated_carbocycles
+
n_saturated_heterocyles
+
n_saturated_rings
+
+
+
+
+
0
+
CHEMBL60745
+
8.787812
+
null
+
245.041526
+
0.400000
+
2
+
1
+
1
+
3
+
13
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
1
+
CHEMBL208599
+
10.585027
+
null
+
298.123676
+
0.388889
+
2
+
2
+
4
+
3
+
21
+
...
+
4.331775
+
2
+
0
+
2
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
2
+
CHEMBL95
+
6.821023
+
4.0
+
198.115698
+
0.307692
+
2
+
2
+
3
+
2
+
15
+
...
+
2.014719
+
1
+
0
+
1
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
3
+
CHEMBL173309
+
7.913640
+
null
+
694.539707
+
0.666667
+
8
+
0
+
2
+
8
+
50
+
...
+
2.803680
+
0
+
0
+
0
+
2
+
0
+
2
+
0
+
0
+
0
+
+
+
4
+
CHEMBL1128
+
6.698970
+
4.0
+
201.092042
+
0.400000
+
2
+
1
+
1
+
3
+
13
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
+
5 rows × 25 columns
+
+
+
+
+
+
+
Training/testing splits
+
Two approaches were used, where one was based purely on max phase split (between max phases null and 4), which was used last time in the decision tree series, and the other one was using the same max phase split but with an ImbalancedLearningRegression method added on top of it.
+
+
+
Preparing training data using max phase split
+
X variable was set up first from the dataframe, and then converted into a NumPy array, which consisted of the number of samples and number of features. This was kept the same as how it was in the decision tree posts.
+
+
+
+
+
+
+Note
+
+
+
+
It’s usually recommended to copy the original data or dataframe before doing any data manipulations to avoid unnecessary changes to the original dataset (this was not used in the decision tree posts, but since I’m going to use the same set of data again I’m doing it here.)
+
+
+
+
# X variables (molecular features)
+# Make a copy of the original dataframe first
+data_mp4 = data.copy()
+# Selecting all max phase 4 compounds
+data_mp4 = data_mp4[data_mp4["max_phase"] ==4]
+print(data_mp4.shape)
+data_mp4.head()
Again, y variable was arranged via the dataframe as well, and converted into a NumPy array. It consisted of the number of samples only as this was the target variable.
Both X and y variables were used to fit the RandomForestRegressor() estimator.
+
+
# n_estimators = 100 by default
+# note: if wanting to use whole dataset - switch off "bootstrap" parameter by using "False"
+rfreg = RandomForestRegressor(max_depth=3, random_state=1, max_features=0.3)
+rfreg.fit(X_mp4, y_mp4)
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Testing data was mainly based on compounds with max phase assigned as “0” or “null” after I renamed it above.
+
+
data_mp_null = data.copy()
+# Selecting all max phase "null" compounds
+data_mp_null = data_mp_null[data_mp_null["max_phase"] =="null"]
+print(data_mp_null.shape)
+data_mp_null.head()
+
+
(466, 25)
+
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
pKi
+
max_phase
+
mw
+
fsp3
+
n_lipinski_hba
+
n_lipinski_hbd
+
n_rings
+
n_hetero_atoms
+
n_heavy_atoms
+
...
+
sas
+
n_aliphatic_carbocycles
+
n_aliphatic_heterocyles
+
n_aliphatic_rings
+
n_aromatic_carbocycles
+
n_aromatic_heterocyles
+
n_aromatic_rings
+
n_saturated_carbocycles
+
n_saturated_heterocyles
+
n_saturated_rings
+
+
+
+
+
0
+
CHEMBL60745
+
8.787812
+
null
+
245.041526
+
0.400000
+
2
+
1
+
1
+
3
+
13
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
1
+
CHEMBL208599
+
10.585027
+
null
+
298.123676
+
0.388889
+
2
+
2
+
4
+
3
+
21
+
...
+
4.331775
+
2
+
0
+
2
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
3
+
CHEMBL173309
+
7.913640
+
null
+
694.539707
+
0.666667
+
8
+
0
+
2
+
8
+
50
+
...
+
2.803680
+
0
+
0
+
0
+
2
+
0
+
2
+
0
+
0
+
0
+
+
+
5
+
CHEMBL102226
+
4.698970
+
null
+
297.152928
+
0.923077
+
3
+
0
+
0
+
5
+
18
+
...
+
2.965170
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
7
+
CHEMBL103873
+
5.698970
+
null
+
269.121628
+
0.909091
+
3
+
0
+
0
+
5
+
16
+
...
+
3.097106
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
+
5 rows × 25 columns
+
+
+
+
+
# Set up X test variable with the same molecular features
+X_mp_test_df = data_mp_null[['mw', 'fsp3', 'n_lipinski_hba', 'n_lipinski_hbd', 'n_rings', 'n_hetero_atoms', 'n_heavy_atoms', 'n_rotatable_bonds', 'n_radical_electrons', 'tpsa', 'qed', 'clogp', 'sas', 'n_aliphatic_carbocycles', 'n_aliphatic_heterocyles', 'n_aliphatic_rings', 'n_aromatic_carbocycles', 'n_aromatic_heterocyles', 'n_aromatic_rings', 'n_saturated_carbocycles', 'n_saturated_heterocyles', 'n_saturated_rings']]
+
+# Convert X test variables from df to arrays
+X_mp_test = X_mp_test_df.to_numpy()
+
+X_mp_test
Training/testing splits using ImbalancedLearningRegression and max phase splits
+
I didn’t really pay a lot of attentions when I was doing data splits in the decision tree series, as my main focus was on building a single tree in order to fully understand and see what could be derived from just one tree. Now, when I reached this series on random forest, I realised I forgot to mention in the last series that data splitting was actually very crucial on model performance and could influence outcome predictions. It could also become quite complicated as more approaches were available to split the data. Also, the way the data was splitted could produce different outcomes.
+
After I’ve splitted the same dataset based on compounds’ max phase assignments in ChEMBL and also fitted the training data on the random forest regressor, I went back and noticed that the training and testing data were very imbalanced and I probably should do something about it before fitting them onto another model.
+
At this stage, I went further to look into whether imbalanced datasets should be addressed in regression tasks, and did a surface search online. So based on common ML concensus, addressing imbalanced datasets were more applicable to classification tasks (e.g. binary labels or multi-class labels), rather than regression problems. However, recent ML research looked into the issue of imbalanced datasets in regression. This blog post mentioned a few studies that looked into this type of problem, and I thought they were very interesting and worth a mention at least. One of them that I’ve looked into was SMOTER, which was based on synthetic minority over-sampling technique (SMOTE)(Chawla et al. 2002), and was named this way because it was basically a SMOTE for regression (hence SMOTER)(Torgo et al. 2013). Synthetic minority over-sampling technique for regression with Gaussian noise (SMOGN)(Kunz 2020) was another technique that was built upon SMOTER, but with Gaussian noises added. This has subsequently led me to ImbalancedLearningRegression library (Wu, Kunz, and Branco 2022), which was a variation of SMOGN. This was the one used on my imbalanced dataset, shown in the section below.
+
A simple flow diagram was drawn below showing the evolution of different techniques when dealing with imbalanced datasets in classification (SMOTE) and regression (SMOTER, SMOGN and ImbalancedLearningRegression):
+
+
+
+
+
flowchart LR
+ A(SMOTE) --> B(SMOTER)
+ B --> C(SMOGN)
+ C --> D(ImbalancedLearningRegression)
+
+
+
+
+
+
+
+
+
GitHub repository for ImbalancedLearningRegression package is available here, with its documentation available here.
+
Also, I just wanted to mention that these were not the only techniques available for treating imbalanced datasets in regression, as there were other ones in the literature and most likely more are being developed currently, but I only had time to cover these here for now.
+
I also would like to mention another really useful open-source resource for treating imbalanced datasets in classifications since I did not use it in this post due to the problem being more of a regression one than a classification one - imbalance-learn library.
+
+
# Original dataset - checking shape again
+print(data.shape)
+data.head()
+
+
(481, 25)
+
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
pKi
+
max_phase
+
mw
+
fsp3
+
n_lipinski_hba
+
n_lipinski_hbd
+
n_rings
+
n_hetero_atoms
+
n_heavy_atoms
+
...
+
sas
+
n_aliphatic_carbocycles
+
n_aliphatic_heterocyles
+
n_aliphatic_rings
+
n_aromatic_carbocycles
+
n_aromatic_heterocyles
+
n_aromatic_rings
+
n_saturated_carbocycles
+
n_saturated_heterocyles
+
n_saturated_rings
+
+
+
+
+
0
+
CHEMBL60745
+
8.787812
+
null
+
245.041526
+
0.400000
+
2
+
1
+
1
+
3
+
13
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
1
+
CHEMBL208599
+
10.585027
+
null
+
298.123676
+
0.388889
+
2
+
2
+
4
+
3
+
21
+
...
+
4.331775
+
2
+
0
+
2
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
2
+
CHEMBL95
+
6.821023
+
4.0
+
198.115698
+
0.307692
+
2
+
2
+
3
+
2
+
15
+
...
+
2.014719
+
1
+
0
+
1
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
3
+
CHEMBL173309
+
7.913640
+
null
+
694.539707
+
0.666667
+
8
+
0
+
2
+
8
+
50
+
...
+
2.803680
+
0
+
0
+
0
+
2
+
0
+
2
+
0
+
0
+
0
+
+
+
4
+
CHEMBL1128
+
6.698970
+
4.0
+
201.092042
+
0.400000
+
2
+
1
+
1
+
3
+
13
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
+
5 rows × 25 columns
+
+
+
+
So my little test on using ImbalancedLearningRegression package started from below.
+
+
iblr_data = data.copy()
+
+# Introducing Gaussian noise for data sampling
+data_gn = iblr.gn(data = iblr_data, y ="pKi", pert =1)
+print(data_gn.shape)
# Followed by max phase split, where max phase 4 = training dataset
+data_gn_mp4 = data_gn[data_gn["max_phase"] ==4]
+data_gn_mp4
+print(data_gn_mp4.shape)
+
+
(7, 25)
+
+
+
+
# Also splitted max phase null compounds = testing dataset
+data_gn_mp_null = data_gn[data_gn["max_phase"] =="null"]
+data_gn_mp_null
+print(data_gn_mp_null.shape)
+
+
(465, 25)
+
+
+
There were several different sampling techniques in ImbalancedLearningRegression package. I’ve only tried random over-sampling, under-sampling and Gaussian noise, but there were also other ones such as SMOTE and ADASYN (in over-sampling technique) or condensed nearest neighbor, Tomeklinks and edited nearest neightbour (in under-sampling technique) that I haven’t used.
+
Random over-sampling actually oversampled the max phase null compounds (sample size increased), while keeping all 10 max phase 4 compounds. Under-sampling removed all of the max phase 4 compounds (which was most likely not the best option, since I was aiming to use them as training compounds), with max phase null compounds also reduced in size too. Due to post length, I did not show the code for random over-sampling and under-sampling, but for people who are interested, I think it would be interesting to test them out.
+
I ended up using Gauissian noise sampling and it reduced max phase 4 compounds slightly, and increased the max phase null compounds a little bit too, which seemed to be the most balanced data sampling at the first try. (Note: as stated from the documentation for ImbalancedLearningRegression package, missing values within features would be removed automatically, I’ve taken care of this in my last series of posts so no difference were observed here.)
+
The change in the distribution of pKi values for the Gaussian noise sampling method between the original and sample-modified datasets could be seen in the kernel density estimate plot below. The modified dataset had a flatter target density curve than the original density plot, which was more concentrated and peaked between pKi values of 6 and 8. The range of pKi values for the ten max phase 4 compounds collected was between 4 and 8.
Then the iblr-gn training data were fitted onto another random forest regressor model.
+
+
# n_estimators = 100 by default
+# note: if wanting to use whole dataset - switch off "bootstrap" parameter by using "False"
+rfreg_gn = RandomForestRegressor(max_depth=3, random_state=1, max_features=0.3)
+rfreg_gn.fit(X_mp4_gn, y_mp4_gn)
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
Modified iblr-gn testing data were also prepared and converted into a NumPy array.
+
+
# Set up X test variable with the same molecular features
+X_mp_gn_test_df = data_gn_mp_null[['mw', 'fsp3', 'n_lipinski_hba', 'n_lipinski_hbd', 'n_rings', 'n_hetero_atoms', 'n_heavy_atoms', 'n_rotatable_bonds', 'n_radical_electrons', 'tpsa', 'qed', 'clogp', 'sas', 'n_aliphatic_carbocycles', 'n_aliphatic_heterocyles', 'n_aliphatic_rings', 'n_aromatic_carbocycles', 'n_aromatic_heterocyles', 'n_aromatic_rings', 'n_saturated_carbocycles', 'n_saturated_heterocyles', 'n_saturated_rings']]
+
+# Convert X test variables from df to arrays
+X_mp_gn_test = X_mp_gn_test_df.to_numpy()
+
+X_mp_gn_test
Using trained model for prediction on testing data
+
Predicting max phase-splitted data only.
+
+
# Predict pKi values for the compounds with "null" max phase
+# using the training model rfreg
+# Uncomment code below to print prediction result
+#print(rfreg.predict(X_mp_test))
+
+# or use:
+y_mp_test = rfreg.predict(X_mp_test)
+
+
Predicting iblr-gn data with max phase splits.
+
+
y_mp_gn_test = rfreg_gn.predict(X_mp_gn_test)
+
+
+
+
+
Scoring and metrics of trained models
+
Checking model accuracy for both training and testing datasets was recommended to take place before moving onto discovering feature importances. A scikit-learn explanation for this could be found in the section on “Permutation feature importance”. So the accuracy scores for the model were shown below.
+
+
# Training set accuracy
+print(f"Random forest regressor training accuracy: {rfreg.score(X_mp4, y_mp4):.2f}")
+
+# Testing set accuracy
+print(f"Random forest regressor testing accuracy: {rfreg.score(X_mp_test, y_mp_test):.2f}")
+
+
Random forest regressor training accuracy: 0.86
+Random forest regressor testing accuracy: 1.00
+
+
+
It looked like both the training and testing accuracies for the random forest regressor model (rfreg) were quite high, meaning that the model was able to remember the molecular features well from the training set (the tiny sample of 10 compounds), and the model was able to apply them to the testing set (which should contain about 400 or so compounds) as well, in order to make predictions on the target value of pKi. This has somewhat confirmed that the model was indeed making predictions, rather than not making any predictions at all, which meant there might be no point in finding out which features were important in the data. Therefore, we could now move onto processing the feature importances to fill in the bigger story i.e. which features were more pivotal towards influencing pKi values of approved drugs targeting acetylcholinesterase (AChE).
+
Similar model accuracy scores were also generated for the iblr-gn modified dataset, which appeared to follow a similar pattern as the max phase-splitted dataset.
+
+
# iblr-Gaussian noise & max phase splitted data
+# Training set accuracy
+print(f"Random forest regressor training accuracy: {rfreg_gn.score(X_mp4_gn, y_mp4_gn):.2f}")
+
+# Testing set accuracy
+print(f"Random forest regressor testing accuracy: {rfreg_gn.score(X_mp_gn_test, y_mp_gn_test):.2f}")
+
+
Random forest regressor training accuracy: 0.79
+Random forest regressor testing accuracy: 1.00
+
+
+
Now, setting up the y_true, which was the acutal pKi values of the testing set, and were converted into a NumPy array too.
I also found out the mean squared error (MSE) between y_true (actual max phase null compounds’ pKi values) and y_pred (predicted max phase null compounds’ pKi values). When MSE was closer to zero, the better the model was, meaning less errors were present.
+
Some references that might help with explaining MSE:
# For max phase splitted dataset only
+mean_squared_error(y_true, y_mp_test)
+
+
2.3988097789702505
+
+
+
When R2 (coefficient of determination) was closer to 1, the better the model is, with a usual range between 0 and 1 (Bruce, Bruce, and Gedeck 2020). If it was negative, then the model might not be performing as well as expected. However, there could be exceptions as other model evaluation methods should also be interpreted together with R2 (a poor R2 might not be wholly indicating it’s a poor model).
+
Some references that might help with understanding R2:
# For max phase splitted dataset only
+r2_score(y_true, y_mp_test)
+
+
-0.16228227953132635
+
+
+
Because the data was re-sampled in a iblr-gn way, the size of array would be different from the original dataset, so here I’ve specifically grabbed pKi values from the iblr-gn modified data to get the actual pKi values for the max phase null compounds.
# MSE for iblr-gn data
+mean_squared_error(y_true_gn, y_mp_gn_test)
+
+
5.7895732090189185
+
+
+
+
# R squared for iblr-gn data
+r2_score(y_true_gn, y_mp_gn_test)
+
+
-0.7425920410726885
+
+
+
Well, it appeared iblr-gn dataset might not offer much advantage than the original max phase splitted method. However, even the max phase splitted method wasn’t that great either, but it might still be interesting to find out which features were important in relation to the pKi values.
+
+
+
+
+
Feature importances
+
There were two types of feature importances available in scikit-learn, which I’ve described below. I’ve also added a Shapley additive explanations (SHAP) approach to this section as well to show different visualisation styles for feature importances on the same set of data.
+
+
+
feature_importances_ attribute from scikit-learn
+
The impurity-based feature importances (also known as Gini importance) were shown below.
+
+
# Compute feature importances on rfreg training model
+feature_imp = rfreg.feature_importances_
+
+
+
# Check what feature_imp looks like (an array)
+feature_imp
I decided to write a function to convert a NumPy array into a plot below as this was also needed in the next section.
+
+
# Function to convert array to df leading to plots
+# - for use in feature_importances_ & permutation_importance
+
+def feat_imp_plot(feat_imp_array, X_df):
+
+"""
+ Function to convert feature importance array into a dataframe,
+ which is then used to plot a bar graph
+ to show the feature importance ranking in the random forest model for the dataset used.
+
+ feat_imp_array is the array obtained from the feature_importances_ attribute,
+ after having a estimator/model fitted.
+
+ X_df is the dataframe for the X variable,
+ where the feature column names will be used in the plot.
+ """
+
+# Convert the feat_imp array into dataframe
+ feat_imp_df = pd.DataFrame(feat_imp_array)
+
+# Obtain feature names via column names of dataframe
+# Rename the index as "features"
+ feature = X_df.columns.rename("features")
+
+# Convert the index to dataframe
+ feature_name_df = feature.to_frame(index =False)
+
+# Concatenate feature_imp_df & feature_name_df
+ feature_df = pd.concat(
+ [feat_imp_df, feature_name_df],
+ axis=1
+ ).rename(
+# Rename the column for feature importances
+ columns = {0: "feature_importances"}
+ ).sort_values(
+# Sort values of feature importances in descending order
+"feature_importances", ascending=False
+ )
+
+# Seaborn bar plot
+ sns.barplot(
+ feature_df,
+ x ="feature_importances",
+ y ="features")
+
+
+
# Testing feat_imp_plot function
+feat_imp_plot(feature_imp, X_mp4_df)
+
+
+
+
+
An alternative way to plot was via Matplotlib directly (note: Seaborn was built based on Matplotlib, so the plots were pretty similar). The code below were probably a bit more straightforward but without axes named and the values were not sorted (only as an example but more code could be added to do this).
There were known issues with the built-in feature_importances_ attribute in scikit-learn. As quoted from scikit-learn on feature importance evaluation:
+
+
… The impurity-based feature importances computed on tree-based models suffer from two flaws that can lead to misleading conclusions. First they are computed on statistics derived from the training dataset and therefore do not necessarily inform us on which features are most important to make good predictions on held-out dataset. Secondly, they favor high cardinality features, that is features with many unique values. Permutation feature importance is an alternative to impurity-based feature importance that does not suffer from these flaws. …
+
+
So I’ve also tried the permutation_importance function (a model-agnostic method).
+
+
perm_result = permutation_importance(rfreg, X_mp_test, y_mp_test, n_repeats=10, random_state=1, n_jobs=2)
+
+# Checking data type of perm_result
+type(perm_result)
+
+
sklearn.utils._bunch.Bunch
+
+
+
It normally returns a dictionary-like objects (e.g. Bunch) with the following 3 attributes:
+
+
importances_mean (mean of feature importances)
+
importances_std (standard deviation of feature importances)
For details on these attributes, this scikit-learnlink will add a bit more explanations.
+
I decided to only use importances_mean for now.
+
+
perm_imp = perm_result.importances_mean
+
+# Confirm it produces an array
+type(perm_imp)
+
+
numpy.ndarray
+
+
+
+
# Using the function feat_imp_plot() on perm_imp result to show plot
+feat_imp_plot(perm_imp, X_mp4_df)
+
+
+
+
+
It generated a different feature importances ranking (if looking at top 6 features), although somewhat similar to the previous one.
+
+
+
+
SHAP approach
+
SHAP values (Lundberg et al. 2020), (Shapley et al. 1953) were used here to provide another way to figure out feature importances. The GitHub repository for this SHAP approach could be accessed here.
+
SHAP’s TreeExplainer() was based on Tree SHAP algorithms (Lundberg et al. 2020), and was used to show and explain feature importances within tree models. It could also be extended to boosted tree models such as LightGBM and XGBoost and also other tree models (as explained by the GitHub repository README.md and its documentation link provided). It was also a model-agnostic method, which could be quite handy.
shap_explainer = shap.TreeExplainer(rfreg)
+
+# X_test needs to be a dataframe (not numpy array)
+# otherwise feature names won't show in plot
+shap_values = shap_explainer.shap_values(X_mp_test_df)
+
+# Horizontal bar plot
+shap.summary_plot(shap_values, X_mp_test_df, plot_type ="bar")
+
+
+
+
+
Dot plot version:
+
+
shap.summary_plot(shap_values, X_mp_test_df)
+
+
+
+
+
Violin plot:
+
+
shap.summary_plot(shap_values, X_mp_test_df, plot_type ="violin")
+
+# Alternative plot option: "layered_violin"
+
+
+
+
+
+
+
+
+
Hyperparameter tuning
+
An example was shown below on tuning the number of trees (n_estimators) used in the random forest model.
+
+
# Function code adapted with thanks from ML Mastery
+# https://machinelearningmastery.com/random-forest-ensemble-in-python/
+
+# ---Evaluate a list of models with different number of trees---
+
+# Define dataset by using the same training dataset as above
+X, y = X_mp4, y_mp4
+
+# Define function to generate a list of models with different no. of trees
+def models():
+# Create empty dictionary (key, value pairs) for models
+ models =dict()
+# Test different number of trees to evaluate
+ no_trees = [50, 100, 250, 500, 1000]
+for n in no_trees:
+ models[str(n)] = RandomForestRegressor(n_estimators=n)
+return models
+
+
+# Define function to evaluate a single model using cross-validation
+def evaluate(model, X, y):
+
+# RepeatedStratifiedKFold usually for binary or multi-class labels
+# - ref link: https://scikit-learn.org/stable/modules/generated/sklearn.model_selection.KFold.html#sklearn.model_selection.KFold
+# so using ReaptedKFold instead
+ cross_val = RepeatedKFold(n_splits=10, n_repeats=15, random_state=1)
+# Run evaluation process & collect cv scores
+# Since estimator/model was based on DecisionTreeRegressor,
+# using neg_mean_squared_error metric
+# n_jobs = -1 meaning using all processors to run jobs in parallel
+ scores = cross_val_score(model, X, y, scoring="neg_mean_squared_error", cv=cross_val, n_jobs=-1)
+return scores
+
+
+# Evaluate results
+# Run models with different RepeatedKFold & different no. of tress
+# with results shown as diff. trees with calculated mean cv scores & std
+
+# Obtain diff. models with diff. trees via models function
+models = models()
+
+# Create empty lists for results & names
+results, names =list(), list()
+
+# Create a for loop to iterate through the list of diff. models
+for name, model in models.items():
+# Run the cross validation scores via evaluate function
+ scores = evaluate(model, X, y)
+# Collect results
+ results.append(scores)
+# Collect names (different no. of trees)
+ names.append(name)
+# Show the average mean squared errors and corresponding standard deviations
+# for each model with diff. no. of trees
+print((name, mean(scores), std(scores)))
+
+
('50', -1.6470594650953017, 1.6444082604560304)
+
+
+
('100', -1.6995136024743887, 1.6797340671624852)
+
+
+
('250', -1.6716290617106646, 1.6236808789148038)
+
+
+
('500', -1.645981936868625, 1.615445700037851)
+
+
+
('1000', -1.6532678610618743, 1.604259597928101)
+
+
+
The negated version of the mean squared error (neg_mean_squared_error) was due to how the scoring parameter source code was written in scikit-learn. It was written this way to take into account of both scoring and loss functions (links provided below for further explanations). All scoring metrics could be accessed here for scikit-learn.
+
Reference links to help with understanding neg_mean_squared_error:
Also, the random forest algorithm was stochastic in nature, meaning that every time hyperparameter tuning took place, it would generate different scores due to random bootstrap sampling. The best approach to evaluate model performance during the cross-validation process was to use the average outcome from several runs of cross-validations, then fit the hyperparameters on a final model, or getting several final models ready and then obtaining the average from these models instead.
+
Below was a version of boxplot plotted using Matplotlib showing the differences in the distributions of the cross validation scores and mean squared errors between different number of trees.
To plot this in Seaborn, I had to prepare the data slightly differently to achieve a different version of the boxplot. Matplotlib was a bit more straightforward to use without these steps.
+
I also used natural sort to sort numerical values (GitHub repository). Otherwise, if using sort_values() only, it would only sort the numbers in lexicographical order (i.e. by first digit only), which was not able to show the tree numbers in ascending order.
+
+
# Combine results & names lists into dataframe
+cv_results = pd.DataFrame(results, index = [names])
+
+
+
# Reset index and rename the number of trees column
+cv_results = cv_results.reset_index().rename(columns={"level_0": "Number_of_trees"})
+
+
+
# Melt the dataframe by number of trees column
+cv_results = cv_results.melt(id_vars="Number_of_trees")
+
+
+
# Sort by the number of trees column
+cv_results = cv_results.sort_values(
+ by="Number_of_trees",
+ key=lambda x: np.argsort(index_natsorted(cv_results["Number_of_trees"]))
+)
The Seaborn boxplot shown should be very similar to the Matplotlib one.
+
Other hyperparameters that could be tuned included:
+
+
tree depths (max_depth)
+
number of samples (max_samples)
+
number of features (max_features) - I didn’t use RDKit to generate molecular features for this post (Datamol version was used instead) which would provide around 209 at least (trying to keep the post at a readable length), but I think this might be a better option when doing cross-validations in model evaluations
+
number of nodes (max_leaf_nodes)
+
+
I’ve decided not to code for these other hyperparameters in the cross-validation step due to length of post (the function code used in cross-validation above could be further adapted to cater for other hyperparameters mentioned here), but they should be looked into if doing full-scale and comprehensive ML using the ensemble random forest algorithm.
+
+
+
+
Final words
+
Random forest was known to be a black-box ML algorithm (Bruce, Bruce, and Gedeck 2020), which was completely different from the white-box ML style revealed in decision tree graphs. Feature importances was therefore crucial to shed some lights and remove some layers of the black-box nature in random forest by showing which features were contributing towards model accuracy by ranking features used to train the model. Cross-validation was also vital to avoid over-fitting (which was more applicable to depth of trees), although in some other cases (e.g. number of trees), it was mentioned that it was unlikely the model would be overfitted. Other options available in scikit-learn ensemble methods that I didn’t get time to try were using voting classifier/regressor and stacking models to reduce biases in models, which might be very useful in other cases.
+
Few things I’ve thought of that I could try to improve what I did here was that I should really look for a different set of testing data, rather than using the max phase splits, which was not that ideal. However, as a lot of us are aware, good drug discovery data are hard to come by (a long-standing and complicated problem), I probably need some luck while looking for a different set of drug discovery data later. Another approach that I could try was that I could use RandomForestClassifier() instead on max phase prediction of these small molecules, rather than making pKi value predictions. This might involve re-labelling the max phases for these compounds into a binary or class labels, then I could use the imbalance-learn package to try and alleviate the problem with imbalanced datasets. Nevertheless, I had some fun working on this post and learnt a lot while doing it, and I hope some of the readers might find this post helpful or informative at least.
+
+
+
+
Acknowledgement
+
I’d like to thank all the authors, developers and contributors who worked towards all of the open-source packages or libraries used in this post. I’d also like to thank all of the other senior cheminformatics and ML practitioners who were sharing their work and knowledge online.
+
+
+
+
+
+
References
+
+Breiman, Leo. 1998. “Arcing Classifier (with Discussion and a Rejoinder by the Author).”The Annals of Statistics 26 (3). https://doi.org/10.1214/aos/1024691079.
+
+Chawla, N. V., K. W. Bowyer, L. O. Hall, and W. P. Kegelmeyer. 2002. “SMOTE: Synthetic Minority over-Sampling Technique.”Journal of Artificial Intelligence Research 16 (June): 321–57. https://doi.org/10.1613/jair.953.
+
+
+Kunz, Nicholas. 2020. SMOGN: Synthetic Minority over-Sampling Technique for Regression with Gaussian Noise (version v0.1.2). PyPI. https://pypi.org/project/smogn/.
+
+
+Lundberg, Scott M., Gabriel Erion, Hugh Chen, Alex DeGrave, Jordan M. Prutkin, Bala Nair, Ronit Katz, Jonathan Himmelfarb, Nisha Bansal, and Su-In Lee. 2020. “From Local Explanations to Global Understanding with Explainable AI for Trees.”Nature Machine Intelligence 2 (1): 2522–5839.
+
+
+Shapley, Lloyd S et al. 1953. “A Value for n-Person Games.”
+
+
+Torgo, Luís, Rita P. Ribeiro, Bernhard Pfahringer, and Paula Branco. 2013. “SMOTE for Regression.” In, 378–89. Springer Berlin Heidelberg. https://doi.org/10.1007/978-3-642-40669-0_33.
+
+
+Wu, Wenglei, Nicholas Kunz, and Paula Branco. 2022. “ImbalancedLearningRegression-a Python Package to Tackle the Imbalanced Regression Problem.” In Joint European Conference on Machine Learning and Knowledge Discovery in Databases, 645–48. Springer.
+
+
]]>
+ Machine learning projects
+ Tree models
+ Pandas
+ Scikit-learn
+ ChEMBL database
+ Python
+ https://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/1_random_forest.html
+ Tue, 21 Nov 2023 11:00:00 GMT
+
+
+ Decision tree
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/1_data_col_prep.html
+
+
Series overview
+
+
Post 1 (this post) - data collection from ChEMBL database using web resource client in Python, with initial data preprocessing
+
Post 2 - more data preprocessing and transformation to reach the final dataset prior to model building
+
Post 3 - estimating experimental errors and building decision tree model using scikit-learn
+
+
+
+
+
Introduction
+
I’ve now come to a stage to do some more machine learning (ML) work after reading a few peer-reviewed papers about ML and drug discovery. It seemed that traditional ML methods were still indispensible performance-wise, and when used in combination with deep learning neural networks, they tend to increase prediction accuracy more. I also haven’t ventured into the practicality and usefulness of large language models in drug discovery yet, but I’m aware work in this area has been started. However, comments from experienced seniors did mention that they are still very much novel and therefore may not be as useful yet. Although by the speed of how things evolve in the so-called “AI” field, this possibly may change very soon. Also from what I can imagine, molecular representations in texts or strings are not quite the same as natural human language texts, since there are a lot of other chemistry-specific features to consider, e.g. chiralities, aromaticities and so on. Because of this, I’m sticking with learning to walk first by trying to cover conventional ML methods in a more thorough way, before trying to run in the deep learning zone.
+
So this leads to this series of posts (3 in total) about decision tree. Previously, I’ve only lightly touched on a commonly used classifier algorithm, logistic regression, as the first series in the ML realm. Reflecting back, I think I could’ve done a more thorough job during the data preparation stage. So this would be attempted this time. The data preparation used here was carried out with strong reference to the materials and methods section in this paper (Tilborg, Alenicheva, and Grisoni 2022), which was one of the papers I’ve read. There are probably other useful methods out there, but this paper made sense to me, so I’ve adopted a few of their ways of doing things during data preprocessing.
+
+
+
+
Data retrieval
+
This time I decided to try something new which was to use the ChEMBL web resource client to collect data (i.e. not by direct file downloads from ChEMBL website, although other useful way could be through SQL queries, which is also on my list to try later). I found this great online resource about fetching data this way from the TeachOpenCADD talktorial on compound data acquisition. The data retrieval workflow used below was mainly adapted from this talktorial with a few changes to suit the selected dataset and ML model.
+
The web resource client was supported by the ChEMBL group and was based on a Django QuerySet interface. Their GitHub repository might explain a bit more about it, particularly the Jupyter notebook link provided in the repository would help a lot regarding how to write code to search for specific data.
+
To do this, a few libraries needed to be loaded first.
+
+
# Import libraries
+# Fetch data through ChEMBL web resource client
+from chembl_webresource_client.new_client import new_client
+
+# Dataframe library
+import pandas as pd
+
+# Progress bar
+from tqdm import tqdm
+
+
To see what types of data were provided by ChEMBL web resource client, run the following code and refer to ChEMBL documentations to find out what data were embedded inside different data categories. Sometimes, it might not be that straight forward and some digging would be required (I went back to this step below to find the “data_validity_comment” when I was trying to do some compound sanitisations actually).
+
+
+
+
+
+
+Note
+
+
+
+
The link provided above also talked about other useful techniques for data checks in the ChEMBL database - a very important step to do during data preprocessing, which was also something I was trying to cover and achieve as much as possible in this post.
+
+
+
+
available_resources = [resource for resource indir(new_client) ifnot resource.startswith('_')]
+print(available_resources)
Resource objects were created to enable API access as suggested by the talktorial.
+
+
# for targets (proteins)
+targets_api = new_client.target
+
+# for bioactivities
+bioact_api = new_client.activity
+
+# for assays
+assay_api = new_client.assay
+
+# for compounds
+cpd_api = new_client.molecule
+
+
Checked object type for one of these API objects (e.g. bioactivity API object).
+
+
type(bioact_api)
+
+
chembl_webresource_client.query_set.QuerySet
+
+
+
+
+
+
Fetching target data
+
A protein target e.g. acetylcholinesterase was randomly chosen by using UniProt to look up the protein UniProt ID.
+
+
# Specify Uniprot ID for acetylcholinesterase
+uniprot_id ="P22303"
+
+# Get info from ChEMBL about this protein target,
+# with selected features only
+targets = targets_api.get(target_components__accession = uniprot_id).only(
+"target_chembl_id",
+"organism",
+"pref_name",
+"target_type"
+)
+
+
The query results were stored in a “targets” object, which was a QuerySet with lazy data evaluation only, meaning it would only react when there was a request for the data. Therefore, to see the results, the “targets” object was then read through Pandas DataFrame.
+
+
# Read "targets" with Pandas
+targets = pd.DataFrame.from_records(targets)
+targets
+
+
+
+
+
+
+
+
+
organism
+
pref_name
+
target_chembl_id
+
target_type
+
+
+
+
+
0
+
Homo sapiens
+
Acetylcholinesterase
+
CHEMBL220
+
SINGLE PROTEIN
+
+
+
1
+
Homo sapiens
+
Acetylcholinesterase
+
CHEMBL220
+
SINGLE PROTEIN
+
+
+
2
+
Homo sapiens
+
Cholinesterases; ACHE & BCHE
+
CHEMBL2095233
+
SELECTIVITY GROUP
+
+
+
+
+
+
+
Selected the first protein target from this dataframe.
+
+
# Save the first protein in the dataframe
+select_target = targets.iloc[0]
+select_target
+
+
organism Homo sapiens
+pref_name Acetylcholinesterase
+target_chembl_id CHEMBL220
+target_type SINGLE PROTEIN
+Name: 0, dtype: object
+
+
+
Then saved the selected ChEMBL ID for the first protein (to be used later).
Checked total rows and columns in the bioactivities dataframe.
+
+
bioact_df.shape
+
+
(706, 15)
+
+
+
+
+
Preprocess bioactivity data
+
When I reached the second half of data preprocessing, an alarm bell went off regarding using half maximal inhibitory concentration (IC50) values in ChEMBL. I remembered reading recent blog posts by Greg Landrum about using IC50 and inhibition constant (Ki) values from ChEMBL. A useful open-access paper (Kalliokoski et al. 2013) from 2013 also looked into this issue about using mixed IC50 data in ChEMBL, and provided a thorough overview about how to deal with situations like this. There was also another paper (Kramer et al. 2012) on mixed Ki data from the same author group in 2012 that touched on similar issues.
+
To summarise both the paper about IC50 and blog posts mentioned above:
+
+
it would be the best to check the details of assays used to test the compounds to ensure they were aligned and not extremely heterogeneous, since IC50 values were very assay-specific, and knowing that these values were extracted from different papers from different labs all over the world, mixing them without knowing was definitely not a good idea
+
the slightly better news was that it was more likely okay to combine Ki values for the same protein target from ChEMBL as they were found to be adding less noise to the data (however ideally similar data caution should also apply)
+
it was also possible to mix Ki values with IC50 values, but the data would need to be corrected via using a conversion factor of 2.0 to convert Ki values to IC50 values (note: I also wondered if this needed to be re-looked again since this paper was published 10 years ago…)
+
+
Because of this, I decided to stick with Ki values only for now before adding more complexities as I wasn’t entirely confident about mixing IC50 values with Ki values yet. Firstly, I checked for all types of units being used in bioact_df. There were numerous different units and formats, which meant they would need to be converted to nanomolar (nM) units first.
It looked like there were duplicates of columns on units and values, so the “units” and “value” columns were removed and “standard_units” and “standard_value” columns were kept instead. Also, “type” column was dropped as there were already a “standard_type” column.
+
+
+
+
+
+
+Note
+
+
+
+
Differences between “type” and “standard_type” columns were mentioned by this ChEMBL blog post.
Then the next step was taking care of any missing entries by removing them in the first place. I excluded “data_validity_comment” column here as this was required to check if there were any unusual activity data e.g. excessively low or high Ki values. A lot of the compounds in this column probably had empty cells or “None”, which ensured that there were no particular alarm bells to the extracted bioactivity data.
+
+
bioact_df.dropna(subset = ["activity_id", "assay_chembl_id", "assay_description", "assay_type", "molecule_chembl_id", "relation", "standard_type", "standard_units", "standard_value", "target_chembl_id", "target_organism"], axis =0, how ="any", inplace =True)
+# Check number of rows and columns again (in this case, there appeared to be no change for rows)
+bioact_df.shape
+
+
(706, 12)
+
+
+
Since all unique units inside the “units” and “values” columns were checked previously, I’d done the same for the “standard_units” column to see the ones recorded in it.
One final check on the number of columns and rows after preprocessing the bioactivity dataframe.
+
+
bioact_df.shape
+
+
(540, 12)
+
+
+
There were a total of 12 columns with 540 rows of data left in the bioactivity dataframe.
+
+
+
+
+
Fetching assay data
+
The assay data was added after I went through the rest of the data preprocessing and also after remembering to check on the confidence scores for assays used in the final data collected (to somewhat assess assay-to-target relationships). This link from ChEMBL explained what the confidence score meant.
+
+
assays = assay_api.filter(
+# Use the previously saved target ChEMBL ID
+ target_chembl_id = chembl_id,
+# Binding assays only as before
+ assay_type ="B"
+).only(
+"assay_chembl_id",
+"confidence_score"
+)
+
+
Placing the fetched assay data into a Pandas DataFrame.
It looked like the lowest confidence score for this particular protein target in binding assays was at 8, with others sitting at 9 (the highest). There were 452 assays with confidence score of 8.
+
+
# Some had score of 8 - find out which ones
+assays_df[assays_df["confidence_score"] ==8]
+
+
+
+
+
+
+
+
+
assay_chembl_id
+
confidence_score
+
+
+
+
+
0
+
CHEMBL634034
+
8
+
+
+
1
+
CHEMBL642512
+
8
+
+
+
2
+
CHEMBL642513
+
8
+
+
+
3
+
CHEMBL642514
+
8
+
+
+
4
+
CHEMBL642515
+
8
+
+
+
...
+
...
+
...
+
+
+
1141
+
CHEMBL3887379
+
8
+
+
+
1142
+
CHEMBL3887855
+
8
+
+
+
1143
+
CHEMBL3887947
+
8
+
+
+
1144
+
CHEMBL3888161
+
8
+
+
+
1874
+
CHEMBL5058677
+
8
+
+
+
+
452 rows × 2 columns
+
+
+
+
+
+
+
Combining bioactivity & assay data
+
The key was to combine the bioactivity and assay data along the “assay_chembl_id” column.
I actually came back to this step to relax the confidence score limit to include all the 8s as well as the 9s (otherwise previously I tried only using assays with score of 9), so that donepezil and galantamine could be included in the dataset as well (the purpose of this would be clearer in post 3 when building the model).
+
+
+
+
Fetching compound data
+
While having identified the protein target, obtained the bioactivity data, and also the assay data, this next step was to fetch the compound data. This could be done by having the ChEMBL IDs available in the bioactivity dataset.
Here, the same step was applied where the compound QuerySet object was converted into a Pandas dataframe. However, the compound data extracted here might take longer than the bioactivity one. One way to monitor progress was through using tqdm package.
Removing any missing entries in the compound data (excluding the “max_phase” column as it was needed during the model training/testing part in post 3 - note: “None” entries meant they were preclinical molecules so not assigned with a max phase yet).
+
+
cpds_df.dropna(subset = ["molecule_chembl_id", "molecule_structures"], axis =0, how ="any", inplace =True)
+
+# Check columns & rows in df
+cpds_df.shape
Ideally, only the compounds with canonical SMILES would be kept. Checking for the types of molecular representations used in the “molecule_structures” column of the compound dataset.
+
+
# Randomly choosing the 2nd entry as example
+cpds_df.iloc[1].molecule_structures.keys()
There were 4 types: “canonical_smiles”, “molfile”, “standard_inchi” and “standard_inchi_key”.
+
+
# Create an empty list to store the canonical smiles
+can_smiles = []
+
+# Create a for loop to loop over each row of data,
+# searching for only canonical_smiles to append to the created list
+for i, cpd in cpds_df.iterrows():
+try:
+ can_smiles.append(cpd["molecule_structures"]["canonical_smiles"])
+exceptKeyError:
+ can_smiles.append(None)
+
+# Create a new df column with name as "smiles",
+# which will store all the canonical smiles collected from the list above
+cpds_df["smiles"] = can_smiles
+
+
Check the compound dataframe quickly to see if a new column for SMILES has been created.
+
+
cpds_df.head(3)
+
+
+
+
+
+
+
+
+
max_phase
+
molecule_chembl_id
+
molecule_structures
+
smiles
+
+
+
+
+
0
+
None
+
CHEMBL28
+
{'canonical_smiles': 'O=c1cc(-c2ccc(O)cc2)oc2c...
+
O=c1cc(-c2ccc(O)cc2)oc2cc(O)cc(O)c12
+
+
+
1
+
3.0
+
CHEMBL50
+
{'canonical_smiles': 'O=c1c(O)c(-c2ccc(O)c(O)c...
+
O=c1c(O)c(-c2ccc(O)c(O)c2)oc2cc(O)cc(O)c12
+
+
+
2
+
None
+
CHEMBL8320
+
{'canonical_smiles': 'O=C1C=CC(=O)C=C1', 'molf...
+
O=C1C=CC(=O)C=C1
+
+
+
+
+
+
+
Once confirmed, the old “molecule_structures” column was then removed.
Clearly, the column that existed in both dataframes was the “molecule_chembl_id” column.
+
The next step was to combine or merge both datasets.
+
+
# Create a final dataframe that will contain both bioactivity and compound data
+dtree_df = pd.merge(
+ bioact_assay_df[["molecule_chembl_id","Ki", "units", "data_validity_comment"]],
+ cpds_df,
+ on ="molecule_chembl_id",
+)
+
+dtree_df.head(3)
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
Ki
+
units
+
data_validity_comment
+
max_phase
+
smiles
+
+
+
+
+
0
+
CHEMBL11805
+
0.104
+
nM
+
Potential transcription error
+
None
+
COc1ccccc1CN(C)CCCCCC(=O)N(C)CCCCCCCCN(C)C(=O)...
+
+
+
1
+
CHEMBL60745
+
1.630
+
nM
+
None
+
None
+
CC[N+](C)(C)c1cccc(O)c1.[Br-]
+
+
+
2
+
CHEMBL208599
+
0.026
+
nM
+
None
+
None
+
CCC1=CC2Cc3nc4cc(Cl)ccc4c(N)c3[C@@H](C1)C2
+
+
+
+
+
+
+
Shape of the final dataframe was checked.
+
+
print(dtree_df.shape)
+
+
(540, 6)
+
+
+
Saving a copy of the merged dataframe for now to avoid re-running the previous code repeatedly, and also to be ready for second-half of the data preprocessing work, which will be in post 2.
+
+
dtree_df.to_csv("ache_chembl.csv")
+
+
+
+
+
+
+
References
+
+Kalliokoski, Tuomo, Christian Kramer, Anna Vulpetti, and Peter Gedeck. 2013. “Comparability of Mixed IC50 Data A Statistical Analysis.” Edited by Andrea Cavalli. PLoS ONE 8 (4): e61007. https://doi.org/10.1371/journal.pone.0061007.
+
+
+Kramer, Christian, Tuomo Kalliokoski, Peter Gedeck, and Anna Vulpetti. 2012. “The Experimental Uncertainty of Heterogeneous Public Ki Data.”Journal of Medicinal Chemistry 55 (11): 5165–73. https://doi.org/10.1021/jm300131x.
+
+
+Tilborg, Derek van, Alisa Alenicheva, and Francesca Grisoni. 2022. “Exposing the Limitations of Molecular Machine Learning with Activity Cliffs.”Journal of Chemical Information and Modeling 62 (23): 5938–51. https://doi.org/10.1021/acs.jcim.2c01073.
+
+
]]>
+ Machine learning projects
+ Tree models
+ Data preprocessing
+ Pandas
+ ChEMBL database
+ Python
+ https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/1_data_col_prep.html
+ Mon, 18 Sep 2023 12:00:00 GMT
+
+
+ Decision tree
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/2_data_prep_tran.html
+
+
Data source
+
The data used in this post 2 for data preprocessing was extracted from ChEMBL database by using ChEMBL web resource client in Python. The details of all the steps taken to reach the final .csv file could be seen in post 1.
+
+
+
+
Checklist for preprocessing ChEMBL compound data
+
Below was a checklist summary for post 1 and post 2 (current post), and was highly inspired by this journal paper (Tilborg, Alenicheva, and Grisoni 2022) and also ChEMBL’s FAQ on “Assay and Activity Questions”.
+
Note: not an exhaustive list, only a suggestion from my experience working on this series, may need to tailor to different scenarios
+
For molecular data containing chemical compounds, check for:
+
+
duplicates
+
missing values
+
salts or mixture
+
+
Check the consistency of structural annotations:
+
+
molecular validity
+
molecular sanity
+
charge standardisation
+
stereochemistry
+
+
Check the reliability of reported experimental values (e.g. activity values like IC50, Ki, EC50 etc.):
+
+
annotated validity (data_validity_comment)
+
presence of outliers
+
confidence score (assays)
+
standard deviation of multiple entries (if applicable)
+
+
+
+
+
Import libraries
+
+
# Import all libraries used
+import pandas as pd
+import math
+from rdkit.Chem import Descriptors
+import datamol as dm
+# tqdm library used in datamol's batch descriptor code
+from tqdm import tqdm
+import mols2grid
+
+
+
+
+
Re-import saved data
+
Re-imported the partly preprocessed data from the earlier post.
There was an extra index column (named “Unnamed: 0”) here, which was likely inherited from how the .csv file was saved with the index already in place from part 1, so this column was dropped for now.
From the above quick statistical summary and also the code below to find the minimum Ki value, it confirmed there were no zero Ki values recorded.
+
+
dtree_df["Ki"].min()
+
+
0.0017
+
+
+
Now the part about converting the Ki values to pKi values, which were the negative logs of Ki in molar units (a PubChem example might help to explain it a little). The key to understand pKi here was to treat pKi similarly to how we normally understand pH for our acids and bases. The formula to convert Ki to pKi for nanomolar (nM) units was:
Applying the calc_pKi function to convert all rows of the compound dataset for the “Ki” column.
+
+
# Create a new column for pKi
+# Apply calc_pKi function to data in Ki column
+dtree_df["pKi"] = dtree_df.apply(lambda x: calc_pKi(x.Ki), axis =1)
+
+
The dataframe would now look like this, with a new pKi column (scroll to the very right to see it).
+
+
dtree_df.head(3)
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
Ki
+
units
+
data_validity_comment
+
max_phase
+
smiles
+
pKi
+
+
+
+
+
0
+
CHEMBL11805
+
0.104
+
nM
+
Potential transcription error
+
NaN
+
COc1ccccc1CN(C)CCCCCC(=O)N(C)CCCCCCCCN(C)C(=O)...
+
9.982967
+
+
+
1
+
CHEMBL60745
+
1.630
+
nM
+
NaN
+
NaN
+
CC[N+](C)(C)c1cccc(O)c1.[Br-]
+
8.787812
+
+
+
2
+
CHEMBL208599
+
0.026
+
nM
+
NaN
+
NaN
+
CCC1=CC2Cc3nc4cc(Cl)ccc4c(N)c3[C@@H](C1)C2
+
10.585027
+
+
+
+
+
+
+
+
+
+
Plan other data preprocessing steps
+
For a decision tree model, a few more molecular descriptors were most likely needed rather than only Ki or pKi and SMILES, since I’ve now arrived at the step of planning other preprocessing steps. One way to do this could be through computations based on canonical SMILES of compounds by using RDKit, which would give the RDKit 2D descriptors. In this single tree model, I decided to stick with only RDKit 2D descriptors for now, before adding on fingerprints (as a side note: I have very lightly touched on generating fingerprints in this earlier post - “Molecular similarities in selected COVID-19 antivirals” in the subsection on “Fingerprint generator”).
+
At this stage, a compound sanitisation step should also be applied to the compound column before starting any calculations to rule out compounds with questionable chemical validities. RDKit or Datamol (a Python wrapper library built based on RDKit) was also capable of doing this.
+
I’ve added a quick step here to convert the data types of “smiles” and “data_validity_comment” columns to string (in case of running into problems later).
There were 3 different types of data validity comments here, which were “Potential transcription error”, “NaN” and “Outside typical range”. So, this meant compounds with comments as “Potential transcription error” and “Outside typical range” should be addressed first.
+
+
# Find out number of compounds with "outside typical range" as data validity comment
+dtree_df_err = dtree_df[dtree_df["data_validity_comment"] =="Outside typical range"]
+print(dtree_df_err.shape)
+dtree_df_err.head()
+
+
(58, 7)
+
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
Ki
+
units
+
data_validity_comment
+
max_phase
+
smiles
+
pKi
+
+
+
+
+
111
+
CHEMBL225198
+
0.0090
+
nM
+
Outside typical range
+
NaN
+
O=C(CCc1c[nH]c2ccccc12)NCCCCCCCNc1c2c(nc3cc(Cl...
+
11.045757
+
+
+
114
+
CHEMBL225021
+
0.0017
+
nM
+
Outside typical range
+
NaN
+
O=C(CCCc1c[nH]c2ccccc12)NCCCCCNc1c2c(nc3cc(Cl)...
+
11.769551
+
+
+
118
+
CHEMBL402976
+
313700.0000
+
nM
+
Outside typical range
+
NaN
+
CN(C)CCOC(=O)Nc1ccncc1
+
3.503485
+
+
+
119
+
CHEMBL537454
+
140200.0000
+
nM
+
Outside typical range
+
NaN
+
CN(C)CCOC(=O)Nc1cc(Cl)nc(Cl)c1.Cl
+
3.853252
+
+
+
120
+
CHEMBL3216883
+
316400.0000
+
nM
+
Outside typical range
+
NaN
+
CN(C)CCOC(=O)Nc1ccncc1Br.Cl.Cl
+
3.499764
+
+
+
+
+
+
+
There were a total of 58 compounds with Ki outside typical range.
With the other comment for potential transciption error, there seemed to be only one compound here.
+
These compounds with questionable Ki values were removed, as they could be potential sources of errors in ML models later on (error trickling effect). One of the ways to filter out data was to fill the empty cells within the “data_validity_comment” column first, so those ones to be kept could be selected.
+
+
# Fill "NaN" entries with an actual name e.g. none
+dtree_df["data_validity_comment"].fillna("none", inplace=True)
+dtree_df.head()
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
Ki
+
units
+
data_validity_comment
+
max_phase
+
smiles
+
pKi
+
+
+
+
+
0
+
CHEMBL11805
+
0.104
+
nM
+
Potential transcription error
+
NaN
+
COc1ccccc1CN(C)CCCCCC(=O)N(C)CCCCCCCCN(C)C(=O)...
+
9.982967
+
+
+
1
+
CHEMBL60745
+
1.630
+
nM
+
none
+
NaN
+
CC[N+](C)(C)c1cccc(O)c1.[Br-]
+
8.787812
+
+
+
2
+
CHEMBL208599
+
0.026
+
nM
+
none
+
NaN
+
CCC1=CC2Cc3nc4cc(Cl)ccc4c(N)c3[C@@H](C1)C2
+
10.585027
+
+
+
3
+
CHEMBL95
+
151.000
+
nM
+
none
+
4.0
+
Nc1c2c(nc3ccccc13)CCCC2
+
6.821023
+
+
+
4
+
CHEMBL173309
+
12.200
+
nM
+
none
+
NaN
+
CCN(CCCCCC(=O)N(C)CCCCCCCCN(C)C(=O)CCCCCN(CC)C...
+
7.913640
+
+
+
+
+
+
+
Filtered out only the compounds with nil data validity comments.
This preprocessing molecules tutorial and reference links provided by Datamol were very informative, and the preprocess function code by Datamol was used below. Each step of fix_mol(), sanitize_mol() and standardize_mol() was explained in this tutorial. I think the key was to select preprocessing options required to fit the purpose of the ML models, and the more experiences in doing this, the more likely it will help with the preprocessing step.
+
+
# _preprocess function to sanitise compounds - adapted from datamol.io
+
+smiles_column ="smiles"
+
+dm.disable_rdkit_log()
+
+def _preprocess(row):
+# Convert each compound to a RDKit molecule in the smiles column
+ mol = dm.to_mol(row[smiles_column], ordered=True)
+# Fix common errors in the molecules
+ mol = dm.fix_mol(mol)
+# Sanitise the molecules
+ mol = dm.sanitize_mol(mol, sanifix=True, charge_neutral=False)
+# Standardise the molecules
+ mol = dm.standardize_mol(
+ mol,
+# Switch on to disconnect metal ions
+ disconnect_metals=True,
+ normalize=True,
+ reionize=True,
+# Switch on "uncharge" to neutralise charges
+ uncharge=True,
+# Taking care of stereochemistries of compounds
+ stereo=True,
+ )
+
+# Added a new column below for RDKit molecules
+ row["rdkit_mol"] = dm.to_mol(mol)
+ row["standard_smiles"] = dm.standardize_smiles(dm.to_smiles(mol))
+ row["selfies"] = dm.to_selfies(mol)
+ row["inchi"] = dm.to_inchi(mol)
+ row["inchikey"] = dm.to_inchikey(mol)
+return row
+
+
Then the compound sanitisation function was applied to the dtree_df.
In this case, I tried using the preprocessing function without adding parallelisation, the whole process wasn’t very long (since I had a small dataset), and was done within a minute or so.
+
Also, as a sanity check on the sanitised compounds in dtree_san_df, I just wanted to see if I could display all compounds in this dataframe as 2D images. I also had a look through each page just to see if there were any odd bonds or anything strange in general.
+
+
# Create a list to store all cpds in dtree_san_df
+mol_list = dtree_san_df["rdkit_mol"]
+# Convert to list
+mol_list =list(mol_list)
+# Check data type
+type(mol_list)
+# Show 2D compound structures in grids
+mols2grid.display(mol_list)
+
+
+
+
+
+
+
+
+
+
+
Detect outliers
+
Plotting a histogram to see the distribution of pKi values first.
I read a bit about Dixon’s Q test and realised that there were a few required assumptions prior to using this test, and the current dataset used here (dtree_san_df) might not fit the requirements, which were:
+
+
normally distributed data
+
a small sample size e.g. between 3 and 10, which was originally stated in this paper (Dean and Dixon 1951).
dtree_san_df.boxplot(column ="pKi")
+
+# the boxplot version below shows a blank background
+# rather than above version with horizontal grid lines
+#dtree_san_df.plot.box(column = "pKi")
+
+
<AxesSubplot: >
+
+
+
+
+
+
I also used Pandas’ built-in boxplot in addition to the histogram to show the possible outliers within the pKi values. Clearly, the outliers for pKi values appeared to be above 10. I also didn’t remove these outliers completely due to the dataset itself wasn’t quite in a Gaussian distribution (they might not be true outliers).
+
+
+
+
Calculate RDKit 2D molecular descriptors
+
I’ve explored a few different ways to compute molecular descriptors, essentially RDKit was used as the main library to do this (there might be other options via other programming languages, but I was only exploring RDKit-based methods via Python for now). A blog post I’ve come across on calculating RDKit 2D molecular descriptors has explained it well, it gave details about how to bundle the functions together in a class (the idea of building a small library yourself to be used in projects was quite handy). I’ve also read RDKit’s documentations and also the ones from Datamol. So rather than re-inventing the wheels of all the RDKit code, I’ve opted to use only a small chunk of RDKit code as a demonstration, then followed by Datamol’s version to compute the 2D descriptors, since there were already a few really well-explained blog posts about this. One of the examples was this useful descriptor calculation tutorial by Greg Landrum.
+
+
+
RDKit code
+
With the lastest format of the dtree_san_df, it already included a RDKit molecule column (named “rdkit_mol”), so this meant I could go ahead with the calculations. So here I used RDKit’s Descriptors.CalcMolDescriptors() to calculate the 2D descriptors - note: there might be more code variations depending on needs, this was just a small example.
+
+
# Run descriptor calculations on mol_list (created earlier)
+# and save as a new list
+mol_rdkit_ls = [Descriptors.CalcMolDescriptors(mol) for mol in mol_list]
+
+# Convert the list into a dataframe
+df_rdkit_2d = pd.DataFrame(mol_rdkit_ls)
+print(df_rdkit_2d.shape)
+df_rdkit_2d.head(3)
+
+
(481, 209)
+
+
+
+
+
+
+
+
+
+
MaxAbsEStateIndex
+
MaxEStateIndex
+
MinAbsEStateIndex
+
MinEStateIndex
+
qed
+
MolWt
+
HeavyAtomMolWt
+
ExactMolWt
+
NumValenceElectrons
+
NumRadicalElectrons
+
...
+
fr_sulfide
+
fr_sulfonamd
+
fr_sulfone
+
fr_term_acetylene
+
fr_tetrazole
+
fr_thiazole
+
fr_thiocyan
+
fr_thiophene
+
fr_unbrch_alkane
+
fr_urea
+
+
+
+
+
0
+
9.261910
+
9.261910
+
0.000000
+
0.000000
+
0.662462
+
246.148
+
230.020
+
245.041526
+
74
+
0
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
1
+
6.509708
+
6.509708
+
0.547480
+
0.547480
+
0.763869
+
298.817
+
279.665
+
298.123676
+
108
+
0
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
2
+
6.199769
+
6.199769
+
0.953981
+
0.953981
+
0.706488
+
198.269
+
184.157
+
198.115698
+
76
+
0
+
...
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
+
3 rows × 209 columns
+
+
+
+
In total, it generated 209 descriptors.
+
+
+
+
Datamol code
+
Then I tested Datamol’s code on this as shown below.
+
+
# Datamol's batch descriptor code for a list of compounds
+dtree_san_df_dm = dm.descriptors.batch_compute_many_descriptors(mol_list)
+print(dtree_san_df_dm.shape)
+dtree_san_df_dm.head(3)
+
+
(481, 22)
+
+
+
+
+
+
+
+
+
+
mw
+
fsp3
+
n_lipinski_hba
+
n_lipinski_hbd
+
n_rings
+
n_hetero_atoms
+
n_heavy_atoms
+
n_rotatable_bonds
+
n_radical_electrons
+
tpsa
+
...
+
sas
+
n_aliphatic_carbocycles
+
n_aliphatic_heterocyles
+
n_aliphatic_rings
+
n_aromatic_carbocycles
+
n_aromatic_heterocyles
+
n_aromatic_rings
+
n_saturated_carbocycles
+
n_saturated_heterocyles
+
n_saturated_rings
+
+
+
+
+
0
+
245.041526
+
0.400000
+
2
+
1
+
1
+
3
+
13
+
2
+
0
+
20.23
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
1
+
298.123676
+
0.388889
+
2
+
2
+
4
+
3
+
21
+
1
+
0
+
38.91
+
...
+
4.331775
+
2
+
0
+
2
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
2
+
198.115698
+
0.307692
+
2
+
2
+
3
+
2
+
15
+
0
+
0
+
38.91
+
...
+
2.014719
+
1
+
0
+
1
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
+
3 rows × 22 columns
+
+
+
+
There were a total of 22 molecular descriptors generated, which seemed more like what I might use for the decision tree model. The limitation with this batch descriptor code was that the molecular features were pre-selected, so if other types were needed, it would be the best to go for RDKit code or look into other Datamol descriptor code that allow users to specify features. The types of descriptors were shown below.
The trickier part for data preprocessing was actually trying to merge, join or concatenate dataframes of the preprocessed dataframe (dtree_san_df) and the dataframe from Datamol’s descriptor code (dtree_san_df_dm).
+
Initially, I tried using all of Pandas’ code of merge/join/concat() dataframes. They all failed to create the correct final combined dataframe with too many rows generated, with one run actually created more than 500 rows (maximum should be 481 rows). One of the possible reasons for this could be that some of the descriptors had zeros generated as results for some of the compounds, and when combining dataframes using Pandas code like the ones mentioned here, they might cause unexpected results (as suggested by Pandas, these code were not exactly equivalent to SQL joins). So I looked into different ways, and while there were no other common columns for both dataframes, the index column seemed to be the only one that correlated both.
+
I also found out after going back to the previous steps that when I applied the compound preprocessing function from Datamol, the index of the resultant dataframe was changed to start from 1 (rather than zero). Because of this, I tried re-setting the index of dtree_san_df first, then dropped the index column, followed by re-setting the index again to ensure it started at zero, which has worked. So now the dtree_san_df would have exactly the same index as the one for dtree_san_df_dm.
+
+
# 1st index re-set
+dtree_san_df = dtree_san_df.reset_index()
+# Drop the index column
+dtree_san_df = dtree_san_df.drop(["index"], axis =1)
+dtree_san_df.head(3)
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
Ki
+
units
+
data_validity_comment
+
max_phase
+
smiles
+
pKi
+
rdkit_mol
+
standard_smiles
+
selfies
+
inchi
+
inchikey
+
+
+
+
+
0
+
CHEMBL60745
+
1.630
+
nM
+
none
+
NaN
+
CC[N+](C)(C)c1cccc(O)c1.[Br-]
+
8.787812
+
<rdkit.Chem.rdchem.Mol object at 0x120080f90>
+
CC[N+](C)(C)c1cccc(O)c1.[Br-]
+
[C][C][N+1][Branch1][C][C][Branch1][C][C][C][=...
+
InChI=1S/C10H15NO.BrH/c1-4-11(2,3)9-6-5-7-10(1...
+
CAEPIUXAUPYIIJ-UHFFFAOYSA-N
+
+
+
1
+
CHEMBL208599
+
0.026
+
nM
+
none
+
NaN
+
CCC1=CC2Cc3nc4cc(Cl)ccc4c(N)c3[C@@H](C1)C2
+
10.585027
+
<rdkit.Chem.rdchem.Mol object at 0x120081cb0>
+
CCC1=CC2Cc3nc4cc(Cl)ccc4c(N)c3[C@@H](C1)C2
+
[C][C][C][=C][C][C][C][=N][C][=C][C][Branch1][...
+
InChI=1S/C18H19ClN2/c1-2-10-5-11-7-12(6-10)17-...
+
QTPHSDHUHXUYFE-KIYNQFGBSA-N
+
+
+
2
+
CHEMBL95
+
151.000
+
nM
+
none
+
4.0
+
Nc1c2c(nc3ccccc13)CCCC2
+
6.821023
+
<rdkit.Chem.rdchem.Mol object at 0x120081700>
+
Nc1c2c(nc3ccccc13)CCCC2
+
[N][C][=C][C][=Branch1][N][=N][C][=C][C][=C][C...
+
InChI=1S/C13H14N2/c14-13-9-5-1-3-7-11(9)15-12-...
+
YLJREFDVOIBQDA-UHFFFAOYSA-N
+
+
+
+
+
+
+
+
# 2nd index re-set
+dtree_san_df = dtree_san_df.reset_index()
+print(dtree_san_df.shape)
+dtree_san_df.head(3)
Checking final dataframe to make sure there were 481 rows (also that index_x and index_y were identical) and also there was an increased number of columns (columns combined from both dataframes). So this finally seemed to work.
+
+
print(dtree_f_df.shape)
+dtree_f_df.head(3)
+
+
(481, 27)
+
+
+
+
+
+
+
+
+
+
index_x
+
molecule_chembl_id
+
pKi
+
max_phase
+
index_y
+
mw
+
fsp3
+
n_lipinski_hba
+
n_lipinski_hbd
+
n_rings
+
...
+
sas
+
n_aliphatic_carbocycles
+
n_aliphatic_heterocyles
+
n_aliphatic_rings
+
n_aromatic_carbocycles
+
n_aromatic_heterocyles
+
n_aromatic_rings
+
n_saturated_carbocycles
+
n_saturated_heterocyles
+
n_saturated_rings
+
+
+
+
+
0
+
0
+
CHEMBL60745
+
8.787812
+
NaN
+
0
+
245.041526
+
0.400000
+
2
+
1
+
1
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
1
+
1
+
CHEMBL208599
+
10.585027
+
NaN
+
1
+
298.123676
+
0.388889
+
2
+
2
+
4
+
...
+
4.331775
+
2
+
0
+
2
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
2
+
2
+
CHEMBL95
+
6.821023
+
4.0
+
2
+
198.115698
+
0.307692
+
2
+
2
+
3
+
...
+
2.014719
+
1
+
0
+
1
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
+
3 rows × 27 columns
+
+
+
+
The two index columns (“index_x” and “index_y”) were removed, which brought out the final preprocessed dataframe.
I then saved this preprocessed dataframe as another file in my working directory, so that it could be used for estimating experimental errors and model building in the next post.
+
+
dtree_f_df.to_csv("ache_2d_chembl.csv")
+
+
+
+
+
Data preprocessing reflections
+
In general, the order of steps could be swapped in a more logical way. The subsections presented in this post bascially reflected my thought processes, as there were some back-and-forths. The whole data preprocessing step was probably still not thorough enough, but I’ve tried to cover as much as I could (hopefully I didn’t go overboard with it…). Also, it might still not be ideal to use Ki values this freely as mentioned in post 1 (noises in data issues).
+
It was mentioned in scikit-learn that for decision tree models, because of its non-parametric nature, there were not a lot of data cleaning required. However, I think that might be domain-specific, since for the purpose of drug discovery, if this step wasn’t done properly, whatever result that came out of the ML model most likely would not work and also would not reflect real-life scenarios. I was also planning on extending this series to add more trees to the model, that is, from one tree (decision tree), to multiple trees (random forests), and then hopefully move on to boosted trees (XGBoost and LightGBM). Therefore, I’d better do this data cleaning step well first to save some time later (if using the same set of data).
+
Next post will be about model building using scikit-learn and also a small part on estimating experimental errors on the dataset - this is going to be in post 3.
+
+
+
+
+
+
References
+
+Dean, R. B., and W. J. Dixon. 1951. “Simplified Statistics for Small Numbers of Observations.”Analytical Chemistry 23 (4): 636–38. https://doi.org/10.1021/ac60052a025.
+
+
+Tilborg, Derek van, Alisa Alenicheva, and Francesca Grisoni. 2022. “Exposing the Limitations of Molecular Machine Learning with Activity Cliffs.”Journal of Chemical Information and Modeling 62 (23): 5938–51. https://doi.org/10.1021/acs.jcim.2c01073.
+
+
]]>
+ Machine learning projects
+ Tree models
+ Data preprocessing
+ ChEMBL database
+ Pandas
+ RDKit
+ Datamol
+ Python
+ https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/2_data_prep_tran.html
+ Mon, 18 Sep 2023 12:00:00 GMT
+
+
+ Decision tree
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/3_model_build.html
+ Post updated on 28th April 2024 - dtreeviz code (under the “Model building” section) were updated to improve the scale of the dtreeviz tree plot
+
+
+
Data source
+
The data used here was extracted from ChEMBL database by using ChEMBL web resource client in Python. The details of all the steps taken to reach the final .csv file could be seen in these earlier posts - post 1 and post 2 (yes, it took quite a while to clean the data, so it was splitted into two posts).
+
+
The final .csv file used to train the model was named, “ache_2d_chembl.csv”
+
The earlier version without any RDKit 2D descriptors calculated was named, “ache_chembl.csv”
+
Both files should be in a GitHub repository called, “ML2-1_decision_tree” or in my blog repository, under “posts” folder (look for “16_ML2-1_Decision_tree” folder)
+
+
+
+
+
Estimate experimental errors
+
This part was about estimating the impact of experimental errors (pKi values) on the predictive machine learning (ML) models. It was also needed to estimate the maximum possible correlation that could be drawn from the dataset prepared from the previous two posts. I supposed it made more sense if this was done prior to building the ML model, so this wouldn’t be forgotten or missed, as we know that real-life is full of many imperfections.
+
This subsection was inspired by Pat Walters’ posts, which have discussed about estimating errors for experimental data with code links available in these posts:
+
+
How Good Could (Should) My Models Be? - a reference paper (Brown, Muchmore, and Hajduk 2009) was mentioned as the simulation basis for estimating the impact of experimental errors on the correlation from a predictive ML model
The pKi column was used in the code below as it contained the experimental values (calculated from measured Ki values, usually derived from countless lab experiments) collected from different scientific literatures or other sources as stated in ChEMBL. The aim was to simulate pKi values with experimental errors added to them.
+
Code used for the rest of the subsection were adapted with thanks from Pat Walters’ “maximum_correlation.ipynb” with my own added comments for further explanations
+
+
# Save exp data (pKi) as an object
+data = dtree["pKi"]
+# Save the object as a list
+data_ls = [data]
+
+# Trial 3-, 5- & 10-fold errors
+for fold in (3, 5, 10):
+# Retrieve error samples randomly from a normal distribution
+# Bewteen 0 and log10 of number-fold
+# for the length of provided data only
+ error = np.random.normal(0, np.log10(fold), len(data))
+ data_ls.append(error + data)
+
+# Convert data_ls to dataframe
+dtree_err = pd.DataFrame(data_ls)
+# Re-align dataframe (switch column header & index)
+dtree_err = dtree_err.transpose()
+# Rename columns
+dtree_err.columns = ["pKi", "3-fold", "5-fold", "10-fold"]
+print(dtree_err.shape)
+dtree_err.head()
+
+
(481, 4)
+
+
+
+
+
+
+
+
+
+
pKi
+
3-fold
+
5-fold
+
10-fold
+
+
+
+
+
0
+
8.787812
+
8.710912
+
9.101193
+
7.471251
+
+
+
1
+
10.585027
+
10.883334
+
10.291557
+
9.455301
+
+
+
2
+
6.821023
+
6.134753
+
6.799967
+
7.122006
+
+
+
3
+
7.913640
+
8.390146
+
7.874722
+
7.209130
+
+
+
4
+
6.698970
+
7.359148
+
7.290723
+
5.770489
+
+
+
+
+
+
+
Melting the created dtree_err so it could be plotted later (noticed there should be an increased number of rows after re-stacking the data).
+
+
# Melt the dtree_err dataframe
+# to make error values in one column (for plotting)
+dtree_err_melt = dtree_err.melt(id_vars ="pKi")
+print(dtree_err_melt.shape)
+dtree_err_melt.head()
+
+
(1443, 3)
+
+
+
+
+
+
+
+
+
+
pKi
+
variable
+
value
+
+
+
+
+
0
+
8.787812
+
3-fold
+
8.710912
+
+
+
1
+
10.585027
+
3-fold
+
10.883334
+
+
+
2
+
6.821023
+
3-fold
+
6.134753
+
+
+
3
+
7.913640
+
3-fold
+
8.390146
+
+
+
4
+
6.698970
+
3-fold
+
7.359148
+
+
+
+
+
+
+
Presenting this in regression plots.
+
Note: There was a matplotlib bug which would always show a tight_layout user warning for FacetGrid plots in seaborn (the lmplot used below). Seaborn was built based on matplotlib so unsurprisingly this occurred (this GitHub issue link might explain it). I have therefore temporarily silenced this user warning for the sake of post publication.
+
+
# To silence the tight-layout user warning
+import warnings
+warnings.filterwarnings("ignore")
+
+# variable = error-fold e.g. 3-fold
+# value = pKi value plus error
+sns.set_theme(font_scale =1.5)
+plot = sns.lmplot(
+ x ="pKi",
+ y ="value",
+ col ="variable",
+ data = dtree_err_melt,
+# alpha = mark’s opacity (low - more transparent)
+# s = mark size (increase with higher number)
+ scatter_kws =dict(alpha =0.5, s =15)
+ )
+title_list = ["3-fold", "5-fold", "10-fold"]
+for i inrange(0, 3):
+ plot.axes[0, i].set_ylabel("pKi + error")
+ plot.axes[0, i].set_title(title_list[i])
+
+
+
+
+
Simulating the impact of error on the correlation between experimental pKi and also pKi with errors (3-fold, 5-fold and 10-fold). R2 calculated using scikit-learn was introduced in the code below.
+
+
# Calculating r2 score (coefficient of determination)
+# based on 1000 trials for each fold
+# note: data = dtree["pKi"]
+
+# Create an empty list for correlation
+cor_ls = []
+for fold in [3, 5, 10]:
+# Set up 1000 trials
+for i inrange(0, 1000):
+ error = np.random.normal(0, np.log10(fold), len(data))
+ cor_ls.append([r2_score(data, data + error), f"{fold}-fold"])
+
+# Convert cor_ls into dataframe
+err_df = pd.DataFrame(cor_ls, columns = ["r2", "fold_error"])
+err_df.head()
This definitely helped a lot with visualising the estimated errors for the experimental Ki values curated in ChEMBL for this specific protein target (CHEMBL220, acetylcholinesterase (AChE)). The larger the error-fold, the lower the R2, and once the experimental error reached 10-fold, we could see an estimated R2 (maximum correlation) with its median sitting below 0.55, indicating a likely poor predictive ML model if it was built based on these data with the estimated 10-fold experimental errors.
+
+
+
+
Check max phase distribution
+
At this stage, I’ve planned to do model training on compounds with max phase 4 (i.e. prescription medicines), so this would somewhat be an attempt to mirror real-life scenarios for the ML prediction model.
+
Max phases were assigned to each ChEMBL-curated compound according to this ChEMBL FAQ link (under the question of “What is max phase?”). As quoted from this ChEMBL FAQ link, a max phase 4 compound means:
+
+
“Approved (4): A marketed drug e.g. AMINOPHYLLINE (CHEMBL1370561) is an FDA approved drug for treatment of asthma.”
+
+
Checking out the actual counts of each max phase group in the dataset.
There was only a very small number of compounds with max phase 4 assigned (a total count of 10, which was also unsurprising since there weren’t many AChE inhibitors used as prescription medications for dementia - some of the well-known examples were donepezil, galantamine and rivastigmine).
+
Filling in actual “null” labels for all “NaN” rows in the “max_phase” columns to help with filtering out these compounds later on.
This was just another sanity check for myself on the dtree dataframe - making sure there weren’t any “NaN” cells in it (so dropping any “NaN” again, even though I might have already done this as one of the steps during data preprocessing).
+
+
dtree.dropna()
+print(dtree.shape)
+dtree.head()
+
+
(481, 25)
+
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
pKi
+
max_phase
+
mw
+
fsp3
+
n_lipinski_hba
+
n_lipinski_hbd
+
n_rings
+
n_hetero_atoms
+
n_heavy_atoms
+
...
+
sas
+
n_aliphatic_carbocycles
+
n_aliphatic_heterocyles
+
n_aliphatic_rings
+
n_aromatic_carbocycles
+
n_aromatic_heterocyles
+
n_aromatic_rings
+
n_saturated_carbocycles
+
n_saturated_heterocyles
+
n_saturated_rings
+
+
+
+
+
0
+
CHEMBL60745
+
8.787812
+
null
+
245.041526
+
0.400000
+
2
+
1
+
1
+
3
+
13
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
1
+
CHEMBL208599
+
10.585027
+
null
+
298.123676
+
0.388889
+
2
+
2
+
4
+
3
+
21
+
...
+
4.331775
+
2
+
0
+
2
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
2
+
CHEMBL95
+
6.821023
+
4.0
+
198.115698
+
0.307692
+
2
+
2
+
3
+
2
+
15
+
...
+
2.014719
+
1
+
0
+
1
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
3
+
CHEMBL173309
+
7.913640
+
null
+
694.539707
+
0.666667
+
8
+
0
+
2
+
8
+
50
+
...
+
2.803680
+
0
+
0
+
0
+
2
+
0
+
2
+
0
+
0
+
0
+
+
+
4
+
CHEMBL1128
+
6.698970
+
4.0
+
201.092042
+
0.400000
+
2
+
1
+
1
+
3
+
13
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
+
5 rows × 25 columns
+
+
+
+
+
+
+
Model building
+
+
Training data based on max phase 4 compounds
+
So here I wanted to separate the collected data by splitting the compounds into two groups based on their assigned max phases. Compounds with max phase 4 were chosen as the training data, and the rest of the compounds with max phases of “null” would be the testing data.
+
+
# Create a df for compounds with max phase 4 only
+dtree_mp4 = dtree[dtree["max_phase"] ==4]
+dtree_mp4
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
pKi
+
max_phase
+
mw
+
fsp3
+
n_lipinski_hba
+
n_lipinski_hbd
+
n_rings
+
n_hetero_atoms
+
n_heavy_atoms
+
...
+
sas
+
n_aliphatic_carbocycles
+
n_aliphatic_heterocyles
+
n_aliphatic_rings
+
n_aromatic_carbocycles
+
n_aromatic_heterocyles
+
n_aromatic_rings
+
n_saturated_carbocycles
+
n_saturated_heterocyles
+
n_saturated_rings
+
+
+
+
+
2
+
CHEMBL95
+
6.821023
+
4.0
+
198.115698
+
0.307692
+
2
+
2
+
3
+
2
+
15
+
...
+
2.014719
+
1
+
0
+
1
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
4
+
CHEMBL1128
+
6.698970
+
4.0
+
201.092042
+
0.400000
+
2
+
1
+
1
+
3
+
13
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
6
+
CHEMBL640
+
6.000000
+
4.0
+
235.168462
+
0.461538
+
4
+
3
+
1
+
4
+
17
+
...
+
1.791687
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
9
+
CHEMBL502
+
7.688246
+
4.0
+
379.214744
+
0.458333
+
4
+
0
+
4
+
4
+
28
+
...
+
2.677222
+
1
+
1
+
2
+
2
+
0
+
2
+
0
+
1
+
1
+
+
+
131
+
CHEMBL481
+
7.296709
+
4.0
+
586.279135
+
0.515152
+
10
+
1
+
7
+
10
+
43
+
...
+
3.632560
+
0
+
4
+
4
+
1
+
2
+
3
+
0
+
2
+
2
+
+
+
133
+
CHEMBL360055
+
4.431798
+
4.0
+
510.461822
+
0.800000
+
6
+
0
+
1
+
6
+
36
+
...
+
3.257653
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
160
+
CHEMBL1025
+
5.221849
+
4.0
+
184.066459
+
1.000000
+
3
+
0
+
0
+
5
+
11
+
...
+
3.345144
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
171
+
CHEMBL659
+
6.522879
+
4.0
+
287.152144
+
0.529412
+
4
+
1
+
4
+
4
+
21
+
...
+
4.226843
+
1
+
2
+
3
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
180
+
CHEMBL1200970
+
4.607303
+
4.0
+
348.142697
+
0.368421
+
2
+
0
+
3
+
4
+
23
+
...
+
4.223591
+
0
+
1
+
1
+
2
+
0
+
2
+
0
+
0
+
0
+
+
+
195
+
CHEMBL1677
+
6.995679
+
4.0
+
234.092376
+
0.307692
+
2
+
2
+
3
+
3
+
16
+
...
+
3.218715
+
1
+
0
+
1
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
+
10 rows × 25 columns
+
+
+
+
Making sure donepezil and galantamine were in this dtree_mp4 dataframe, so the model training would be based on these medicines and also other max phase 4 AChE inhibitors.
+
The screenshots of both medicines were taken from ChEMBL website:
+
+
+
+Screenshot of donepezil (parent drug form) with its molecule ChEMBL ID
+
+
+
+
+
+Screenshot of galantamine (parent drug form) with its molecule ChEMBL ID
+
+
+
The following regex string check confirmed that these two compounds were in the dtree_mp4 dataframe - row indices 9 and 171 contained these two drugs.
The DecisionTreeRegressor() was fitted on the compounds with max phase 4 as shown below, keeping tree depth at 3 for now to avoid complicating the overall tree graph (the deeper the tree, the more branches - potentially might overfit and create noises for the model).
The following was a dtreeviz version of the decision tree, which actually included the regression plots of different molecular features e.g. clogp versus the target value of pKi. It seemed a bit more intuitive as these plots clearly showed where the threshold cut-offs would be for each feature (molecular descriptors). The GitHub repository link for dtreeviz could be accessed here.
+
+
import dtreeviz
+
+viz = dtreeviz.model(ache_tree_mp4, X_train=X_mp4, y_train=y_mp4, target_name="pKi", feature_names=list(X_mp4_df.columns))
+# Added "scale = 2" to view()
+# to make plot larger in size
+viz.view(scale =2)
+
+
+
+
+
+
+
+
Testing and predicting data based on max phase of null compounds
+
+
# Compounds with max phase as "null"
+dtree_mp_null = dtree[dtree["max_phase"] =="null"]
+print(dtree_mp_null.shape)
+dtree_mp_null.head()
+
+
(466, 25)
+
+
+
+
+
+
+
+
+
+
molecule_chembl_id
+
pKi
+
max_phase
+
mw
+
fsp3
+
n_lipinski_hba
+
n_lipinski_hbd
+
n_rings
+
n_hetero_atoms
+
n_heavy_atoms
+
...
+
sas
+
n_aliphatic_carbocycles
+
n_aliphatic_heterocyles
+
n_aliphatic_rings
+
n_aromatic_carbocycles
+
n_aromatic_heterocyles
+
n_aromatic_rings
+
n_saturated_carbocycles
+
n_saturated_heterocyles
+
n_saturated_rings
+
+
+
+
+
0
+
CHEMBL60745
+
8.787812
+
null
+
245.041526
+
0.400000
+
2
+
1
+
1
+
3
+
13
+
...
+
3.185866
+
0
+
0
+
0
+
1
+
0
+
1
+
0
+
0
+
0
+
+
+
1
+
CHEMBL208599
+
10.585027
+
null
+
298.123676
+
0.388889
+
2
+
2
+
4
+
3
+
21
+
...
+
4.331775
+
2
+
0
+
2
+
1
+
1
+
2
+
0
+
0
+
0
+
+
+
3
+
CHEMBL173309
+
7.913640
+
null
+
694.539707
+
0.666667
+
8
+
0
+
2
+
8
+
50
+
...
+
2.803680
+
0
+
0
+
0
+
2
+
0
+
2
+
0
+
0
+
0
+
+
+
5
+
CHEMBL102226
+
4.698970
+
null
+
297.152928
+
0.923077
+
3
+
0
+
0
+
5
+
18
+
...
+
2.965170
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
7
+
CHEMBL103873
+
5.698970
+
null
+
269.121628
+
0.909091
+
3
+
0
+
0
+
5
+
16
+
...
+
3.097106
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
0
+
+
+
+
5 rows × 25 columns
+
+
+
+
There were 466 compounds with max phase as “null”, meaning they were pre-clinical compounds. This was confirmed through the answer from ChEMBL FAQ link, a max phase of “null” compound means:
+
+
“Preclinical (NULL): preclinical compounds with bioactivity data e.g. is a preclinical compound with bioactivity data that has been extracted from scientific literature. However, the sources of drug and clinical candidate drug information in ChEMBL do not show that this compound has reached clinical trials and therefore the max_phase is set to null.”
+
+
Again, setting up the features for the testing dataset.
Because of the small amount of training data, this might hint at using an ensemble approach in the future, where model averaging would be derived from a bunch of tree models rather than using a single tree model, which was what I did here. The reason I started with one tree was because it was no point in building a multiple-tree model if one had no clue about how one tree was built. To learn as much as possible, I had to dissect the multiple-tree version first to focus on one tree at a time.
+
One thing I’d like to mention was that rivastigmine was not included in the training dataset because it was actually not a pure AChE inhibitor (as it was also a butyrylcholinesterase (BChE) inhibitor), since my focus was purely on AChE at this time, this particular drug was unfortunately excluded. However, I did make sure the other two drugs (donepezil and galantamine) were included in the training dataset. One possible thing to do in the future if I want to improve this was to add BChE as another protein target and perhaps add this as an additional dataset towards the model.
+
As described in the subsection of “Estimate experimental errors”, there were experimental errors of 3-fold, 5-fold and 10-fold estimated based on the provided pKi data. With the prediction model used in this post, the estimated experimental errors would need to be taken into consideration, particularly at the time when the model was being investigated during the model evaluation and validation step (however due to the length of series 2.1 posts, I decided not to add this step yet, but would try to look at this later in the multiple tree model series if this applies).
+
A bit about the last decision tree plot, tentatively clogp (calculated partition coefficient) might be the crucial molecular feature in deciding whether a molecule might be closer to being an AChE inhibitor. Other important molecular features also included the number of aromatic rings, molecular weights, solvent accessible surface area and others (I’ve set the random state to 1 for now, so hopefully the result will be reproducible as I realised my old code without it always generated different tree plots, then all of the sudden I remembered that I forgot to set the random state of the estimator, so this was added).
+
Since the type of AChE inhibitors was not the focus of this series, I won’t go into details about which value of pKi or Ki would lead to the ideal AChE inhibitor (the well-known Cheng-Prusoff equation (Cheng and Prusoff 1973) might also lead to many discussions about Ki and IC50 values). This is because there are at least two types of AChE inhibitors available - reversible and irreversible (Colovic et al. 2013). Donepezil, galantamine and rivastigmine mentioned previously are the commonly known reversible AChE inhibitors. The irreversible type, as the name suggested, is usually used as insecticides or nerve agents. Another reason is that I didn’t go into details checking all of the identities for the 10 max phase 4 compounds used in the training set, as I only really made sure that donepezil and galantamine were included in the 10 molecules. If I were to re-model again purely on reversible AChE inhibitors targeting dementia or Alzheimer’s disease, I think I had to be quite sure of what I was training the model with, i.e. excluding irreversible AChE inhibitors from the training set.
+
However, if our aim was to only find novel AChE inhibitors in a general manner, one of the ways to check post-model building would be to re-run the dataframe again on compounds with max phase as null, including the molecular feature names to find out which compounds were at the predicted threshold cut-off values to see if their corresponding pKi values (note: these preclinical compounds had Ki values extracted from literature sources etc.) would match the predicted ones. One caveat of this method was that there might be pre-existing experimental errors in all the obtained and recorded Ki values, so this might not confirm that the model was truly a good reflection of the real-life scenario. Therefore, at most, this would probably add a very small value during the model evaluation phase.
+
The best way would be to test all of these compounds in the same experimental set-ups, through same experimental steps, and in the same laboratory to find out their respective Ki (or pKi) values. However, this was most likely not very feasible due to various real-life restrictions (the availability of financial and staffing resources). The most likely outcome might be to choose a selected group of compound candidates with the highest possibilities to proceed in the drug discovery pipeline based on past experimental, ML and clinical experiences, and then test them in the ways mentioned here.
+
I also came across a blog post about calculating the prediction intervals of ML prediction models (which mentioned the MAPIE package), but I didn’t quite get time to look into this package yet, and from what I have read in its repository link, it potentially could be quite useful for classification, regression and time-series models.
+
+
+
+
Final words
+
I didn’t think a definite conclusion could be drawn here, as this was only purely from one very simple and single decision tree, so I have named this last part as “final words”, as I felt if I didn’t stop here, this post or series of posts could go on forever or as long it could. The main thing here was to fully understand how one single decision tree was constructed based on hopefully reasonable-ish data (still not the best as I could not rule out all the noises from the data), and then to view the tree visually in different styles of plots. It was also important to understand how this was a white-box ML approach with clear features or descriptions shown to trace where the tree would branch off to reach different final outcomes or targets. This series was really a preamble for the multiple-tree models e.g. random forest and boosted trees, as I have bravely planned to do a series of posts on tree models due to my interests in them, so that might take a while, slowly but hopefully surely.
+
+
+
+
Acknowledgements
+
I’d like to thank all the authors for all the open-source packages used in the series 2.1 posts. I’d also like to thank all the authors of all the blog posts mentioned in this series as well since I’ve learnt a lot from them too.
+
+
+
+
+
+
References
+
+Brown, Scott P., Steven W. Muchmore, and Philip J. Hajduk. 2009. “Healthy Skepticism: Assessing Realistic Model Performance.”Drug Discovery Today 14 (7-8): 420–27. https://doi.org/10.1016/j.drudis.2009.01.012.
+
+
+Cheng, Yung-Chi, and William H. Prusoff. 1973. “Relationship Between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction.”Biochemical Pharmacology 22 (23): 3099–3108. https://doi.org/10.1016/0006-2952(73)90196-2.
+
+
+Colovic, Mirjana B., Danijela Z. Krstic, Tamara D. Lazarevic-Pasti, Aleksandra M. Bondzic, and Vesna M. Vasic. 2013. “Acetylcholinesterase Inhibitors: Pharmacology and Toxicology.”Current Neuropharmacology 11 (3): 315–35. https://doi.org/10.2174/1570159x11311030006.
+
+
]]>
+ Machine learning projects
+ Tree models
+ Pandas
+ Scikit-learn
+ ChEMBL database
+ Python
+ https://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/3_model_build.html
+ Mon, 18 Sep 2023 12:00:00 GMT
+
+
+
+ Molecular visualisation (Molviz) web application
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/15_Molviz/Molviz.html
+ The final deployed app is on Shinyapps.io:
+
+
Link: here or please visit https://jhylin.shinyapps.io/molviz_app/
+
Code: here or please visit https://github.com/jhylin/Molviz_app
+
+
+
+
Background - how the app started
+
Originally I had an idea of incorporating mols2grid library within Shiny for Python web app framework (after seeing an example of a similar app in Streamlit previously). So I worked on a few ideas, but obviously mols2grid was designed to work inside Jupyter Notebook/Lab and Shiny for Python was only out of alpha at that stage so things were still being developed. After a few trials, unfortunately mols2grid wasn’t directly compatible with the Shiny for Python framework at that time (I even wrote a small story about it as a comment to an issue).
+
I then went away to work on another project on molecular scaffolds and left this mini project aside. However, recently I had another idea of trying to build a Shiny for Python app from the scratch (with a focus on cheminformatics or chemical information), so that users in relevant fields can view and save 2D images of small molecules in a web browser environment instead of only inside a Jupyter Notebook/Lab. I also thought to place the Shiny for Python framework to test if it was being used in a more intensive area such as chemistry and drug discovery.
+
Another reason that have triggered this side project was that I came across a comment from an old RDKit blog post from someone asking about how to save compound image as a PNG1 file, since SVG2 version was hard to convert etc. (or something along that line). I thought it should be possible, and this should not be only limited to Jupyter environments only (thinking of people not doing coding at all…), so here we are.
+
+
+
+
About each version of the app
+
I’ll try to explain what each version of the app_x.py script entails, as there are currently several different versions of them inside the repository. The final version is the one called “app.py”, named this way so that it’ll be recognised by rsconnect/Shinyapps.io when deploying the app. By providing some explanations below should also show that it was quite a process to arrive at the final “app.py”, it wasn’t built within a day for sure (at least for me).
+
+
+
app_molviz_v1.py
+
This was the first version that purely provided the ability to show 2D images of the molecules via selecting their corresponding index numbers. The libraries used appeared less aligned and a few tests were run below (some of them commented out during trials). This was the one that I’ve figured out how to make the image appeared in the app.
+
+
+
+
app_molviz_v2.py
+
For the second version, I started thinking about how I would present the app in a simple layout for the end users. The backbone code to support image generations was by using rdkit.Chem.Draw package’s MolToImage() module, which normally returns a PIL3 image object, and also supports atom and bond highlighting. Another useful module that I’ve tried was MolToFile() within the same package, which would generate and save a PNG file for a specified molecule from the dataframe.
+
I then took a bit more time to familiarise myself with some basic PIL image manipulations, and used online resources to formulate code to merge separate PNG images into one table grid-like image - potentially may be useful for substructural or R-group comparisons.
+
I have also added the interactive data table at the end to see how it would fit in with the rest of the app.
+
+
+
+
app_molviz_v3.py
+
The third version mainly dealt with how to segregate and differentiate between highlighting or non-highlighting and also with or without index numbers showing for the compounds in the images. I’ve tried to use a different code for atom labelling this time with thanks to this link. However, there was always an issue of not being able to flip back from with index to without index, since the atom labelling code itself somehow overflows its effect to the rest after labelling the atom indices (presumably this atom labelling code would work great in a Jupyter notebook scenario).
+
+
+
+
app_molviz_v4.py & app_molviz_v5.py
+
Both version 4 and 5 were where I’ve tested using “atomNote” (numbers appear beside atoms) instead of “atomLabel” (numbers replaces atoms directly in structures) to label atoms in molecular structures.
+
An example of the atom labelling code would look like this (replace ‘atomNote’ with ‘atomLabel’ to get different labelling effect):
+
```{python}
+for atom in mols[input.mol()].GetAtoms():
+ atom.SetProp('atomNote', str(atom.GetIdx()))
+```
+
I’ve also started adding introductory texts for the app and edited the layout a bit more.
+
+
+
+
app_molviz_v6_hf.py
+
This was basically the final version of the app, but with code edited to attempt to deploy the app on HuggingFace. The main difference I was testing was on how to store the saved images as Docker was new to me at the time, and then while I was thinking about changing the Dockerfile, there was actually another problem in relation to the cairosvg code. Because of this, I then placed this deployment on hold in order to buy more time to figure out code, and also to try Shinyapps.io to see if this could be deployed.
+
+
+
+
app_molviz_v6.py or app.py
+
This was the last version and was the version used to deploy the app on Shinyapps.io. I had to rename the file as mentioned previously to “app.py” so that the Shinyapps.io servers would recognise this Python script as the script to run the app (otherwise it wouldn’t be deployed successfully, this took me a few tries and to read the log file to figure this out). So it was saved as a separate file, and for any latest text changes in the app I would refer to app.py as the most current app file.
+
The biggest code change was that I ended up not using the MolToImage() or MolToFile() modules, but rather I used rdMolDraw2D module from rdkit.Chem.Draw package. The reason being I’ve noticed the image resolutions weren’t great for the previously used modules (Jupyter notebook environments should not have this problem, as you could simply switch on this line of code by setting it to true like this, IPythonConsole.ipython_useSVG = True). So I resorted to other means and came across this useful link to generate images with better resolutions, and introduced the cairosvg library.
+
So the code was changed and would now use rdMolDraw2D.MolDraw2DSVG() first and add on addAtomIndices from drawOptions() and also DrawMolecule() to highlight substructures. The SVG generated would then be converted to PNG via cairosvg library. The end result produced slightly better image resolutions. Although I’ve found for more structurally complexed molecules, the image size would really need to be quite large to be in the high resolution zone. For compounds with simpler structures, this seemed to be much less of a problem. This was also why I had to have these PNG images blown up this large in the app, to cater for the image resolution aspect.
+
+
+
+
+
Other files
+
+
code_test.py
+
I’m not exactly sure how other data scientists/developers work, but for me since I came from a completely different background and training, I’m used to plan, set up and do experiments to test things I’d like to try, and see where the results will lead me to. So for this in a virtual computer setting, I used the “code_tests.py” to test a lot of different code.
+
If you go into this file, you’ll likely see a lot of RDKit code trials, and I have had a lot of fun doing this since I got to see results straight away when running the code, and learn new code and functions that way. If the end result was not the one I’ve intended, I would go on short journeys to look for answers (surprisingly I didn’t use any generative AI chatbots actually), it would be chosen intuitively as I searched online, but for this particular project, a lot of it was from past RDKit blogs, StackOverflow and random snippets I came across that have given me ideas about solving issues I came across.
+
+
+
+
app_table.py & app_itables.py
+
These two files were trials for incorporating dataframe inside a web app. The difference was that app_table.py was just a data table sitting inside the app, without any other particular features, while app_itables.py utilised a different package called itables, which provided an interactive feature to search for data in the table. The previous post on data source used for this app was presented as an interactive data table embedded inside a Quarto document, the same principle would also apply for a table inside a Jupyter notebook environment.
+
+
+
+
app_sample.py
+
This file was provided by Posit (formerly known as RStudio) from their Python for Shiny app template in HuggingFace as an example script for an app.
+
+
+
+
+
Features of the app
+
There are three main features for this app which allows viewing, saving4 and highlighting substructures of target molecules as PNG image files. I’m contemplating about adding a download feature for image file saving on the deployed app version but because I’m currently using the free version of Shinyapps.io with limited amount of data available, this may be unlikely (also because the app is more of a demonstration really as the focus is not to provide particular data/image downloads).
+
+
+
+
App deployment
+
There were two places I’ve tried so far, which were HuggingFace and Shinyapps.io. As mentioned briefly earlier under the subsection of “app_molviz_v6_hf.py”, it turned out cairosvg code didn’t quite play out as expected. I have so far not returned to fix this yet on HuggingFace, since I’ve managed to deploy the app on Shinyapps.io. I had a feeling I might need to revert back to the older code version with poorer image resolutions, so that was also another reason why I haven’t fixed it yet as I’d prefer to keep the better resolution one (unless someone has better ideas out there).
+
However, deploying the app to Shinyapps.io also wasn’t a smooth ride as well, there were some problems initially. The very first problem I got was being told by the error message that rsconnect-python was only compatible with Python version 3.7, 3.8, 3.9 and 3.10 only. I did some information digging in the Posit community forum, and I think several people mentioned using 3.9 without any problems to deploy their apps. Python version 3.11 definitely did not work at all so please avoid for now if anyone would like to try using Shiny for Python app (unless updated by rsconnect-Python in the future).
+
So I think the ideal app building workflow might be like this:
+
Note: all code examples below are to be run in the command line interface
+
+
Refer to this link provided by Shiny for Python, which details about how to set up the working directory, download the Shiny package and create a virtual environment
+
When creating the virtual environment, use venv which was already built-in within Python (and also as suggested by the Shiny for Python link) and set it to a compatible Python version.
+
+
```{python}
+# To create a venv with a specific Python version e.g. Python 3.9
+python3.9-m venv my_venv_name
+
+# Activate the created venv
+source my_venv_name/bin/activate
+```
+
+
If you’ve accidentally set it to Python 3.11 (like what I did), just deactivate the old venv and re-create another one by using the code above. The code below can be used to deactivate the venv set up in the first place.
+
+
```{python}
+# Deactivate the old venv
+deactivate
+```
+
+
If you had to set up a new venv with a new Python version, and did not want to re-add/install all the packages or libraries used in the older version, save all the pip packages like this code below as a requirements.txt file.
+
+
```{python}
+pip freeze > requirements.txt
+```
+
+
Once the requirements.txt was saved and after the new venv was set up and activated, install all the saved packages used to run the app by using this code.
+
+
```{python}
+pip freeze -r requirements.txt
+```
+
+
Start coding for your app and have fun - don’t forget to save and push the files to your repository.
+
To deploy to Shinyapps.io, follow this link, which explains all the steps. One thing I would like to remind again here is to make sure the app script (i.e. the one with data source, user interface and server code) was saved as “app.py”, so that the rsconnect-python server will recognise it and be able to deploy it to the cloud environment.
+
+
+
+
+
Further improvements of the app
+
There are of course a few things I think could be done to make the app better.
+
+
It may be useful to add a download option as mentioned previously, but for demonstration purpose, I’m leaving it as a “View” only for now, unless I get comments from readers that they’d like to try this. For localhost version, the saving image function should work with files saved to the working directory.
+
It may be even better to use SMARTS5 or SMILES for highlighting compound substructures actually (atom and bond numbering can be a bit tricky, I’ve tried the app myself, and it might not be as straight forward). I’m using atom indices here since I’m using a specific code in RDKit, but perhaps more experienced users for RDKit will know how to make code alterations etc.
+
The app layout could be further optimised for aesthetics e.g. interactive data table could be placed at a different location, and potentially the data table could contain other data such as compound bioassay results to really fit in the structure-activity relationship exploring task.
+
+
+
+
+
Final words
+
The whole idea behind this side project was to show that interested users could use this web app framework to build an interactive app by using their own data. Other useful web app frameworks are also available out there and could potentially be more or equally useful as this one (I’m simply testing out Python for Shiny here since it’s relatively new). In a drug discovery and development setting, this could be useful to make non-coding members understand where the computer side was trying to do, and to possibly assist them during their lab workflows, hoping to add some convenience at least.
This is currently limited to localhost version if running the app.py in IDE such as VS Code, where the saved files can be located in the working directory. The deployed version on Shinyapps.io currently only allow image viewing and structure highlighting only.↩︎
+
SMILES arbitrary target specification↩︎
+
+
]]>
+ Python
+ Datamol
+ Shiny
+ Pandas
+ Polars
+ itables
+ https://jhylin.github.io/Data_in_life_blog/posts/15_Molviz/Molviz.html
+ Wed, 09 Aug 2023 12:00:00 GMT
+
+
+ Molecular visualisation (Molviz) web application
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/15_Molviz/itables.html
+
+
Introduction
+
This time I’m trying to build a web application in the hope to contribute my two cents towards minimising the gap between computational and laboratory sides in a drug discovery (or chemistry-related work) setting. There are definitely many other useful web applications available out there for similar uses, but I guess each one has its own uniqueness and special features.
+
For this app, it is mostly aimed at lab chemists who do not use any computer programming code at all in their work, and would like to quickly view compounds in the lab while working, and also to be able to highlight compound substructures during discussions or brainstorming for ideas during compound synthesis. Importantly, this app can exist outside a Jupyter notebook environment with internet required to access the app.
+
This is also the first part prior to the next post which will showcase the actual app. This part mainly involves some data cleaning but not as a major focus for this post. This is not to say that data cleaning is not important, but instead they are fundamental to any work involving data in order to ensure reasonable data quality, which can then potentially influence decisions or results. I have also collapsed the code sections below to make the post easier to read (to access code used for each section, click on the “Code” links).
+
+
+
+
Code and explanations
+
It was actually surprisingly simple for this first part when I did it - building the interactive table. I came across this on LinkedIn on a random day for a post about itables being integrated with Shiny for Python plus Quarto. It came at the right time because I was actually trying to build this app. I quickly thought about incorporating it with the rest of the app so that users could refer back to the data quickly while visualising compound images. The code and explanations on building an interactive table for dataframes in Pandas and Polars were provided below.
+
To install itables, visit here for instructions and also for other information about supported notebook editors.
+
+
+Code
+
# Import dataframe libraries
+import pandas as pd
+import polars as pl
+
+# Import Datamol
+import datamol as dm
+
+# Import itables
+from itables import init_notebook_mode, show
+init_notebook_mode(all_interactive=True)
+
+
+# Option 1: Reading df_ai.csv as a pandas dataframe
+#df = pd.read_csv("df_ai.csv")
+#df.head
+
+# Option 2: Reading df_ai.csv as a polars dataframe
+df = pl.read_csv("df_ai.csv")
+#df.head()
+
+
+# Below was the code I used in my last post to fix the missing SMILES for neomycin
+# - the version below was edited due to recent updates in Polars
+# Canonical SMILES for neomycin was extracted from PubChem
+# (https://pubchem.ncbi.nlm.nih.gov/compound/Neomycin)
+
+df = df.with_columns([
+ pl.when(pl.col("Smiles").str.lengths() ==0)
+ .then(pl.lit("C1C(C(C(C(C1N)OC2C(C(C(C(O2)CN)O)O)N)OC3C(C(C(O3)CO)OC4C(C(C(C(O4)CN)O)O)N)O)O)N"))
+ .otherwise(pl.col("Smiles"))
+ .keep_name()
+])
+
+#df.head()
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
Polars dataframe library was designed without the index in mind (which is different to Pandas), therefore the itables library did not work on my specific polars dataframe which required an index column to show (note: all other Polars dataframes should work fine with itables without the index column!).
+
However to show row counts in Polars dataframes, we could use with_row_count() that starts the index from 0, and this would show up in a Jupyter environment as usual. A small code example would be like this below.
+
+
+Code
+
# Uncomment below to run
+#df = df.with_row_count()
+
+
+
Then I converted the Polars dataframe into a Pandas one (this could be completely avoided if you started with Pandas actually).
+
+
+Code
+
df = df.to_pandas()
+
+
+
Then I added Datamol’s “_preprocess” function to convert SMILES1 into other molecular representations such as standardised SMILES (pre-processed and cleaned SMILES), SELFIES2, InChI3, InChI keys - just to provide extra information for further uses if needed. The standardised SMILES generated here would then be used for generating the molecule images later (in part 2).
# Saving cleaned df_ai.csv as a new .csv file (for app_image_x.py - script to build the web app)
+# df = pl.from_pandas(df)
+# df.write_csv("df_ai_cleaned.csv", sep = ",")
+
+
+
+
+
Options for app deployment
+
Since I had a lot of fun deploying my previous app in Shinylive last time, I thought I might try the same this time - deploying the Molviz app as a Shinylive app in Quarto. However, it didn’t work as expected, with reason being that RDKit wasn’t written in pure Python (it was written in Python and C++), so there wasn’t a pure Python wheel file available in PyPi - this link may provide some answers relating to this. Essentially, packages or libraries used in the app will need to be compatible with Pyodide in order for the Shinylive app to work. So, the most likely option to deploy this app now would be in Shinyapps.io or HuggingFace as I read about it recently.
+
+
+
+
Next post
+
Code and explanations for the actual Molviz app will be detailed in the next post. To access full code and files used for now, please visit this repository link.
+
+
+
+
+
+
Footnotes
+
+
+
Simplified Molecular Input Line Entry Systems↩︎
+
SELF-referencIng Embedded Strings↩︎
+
International Chemical Identifier↩︎
+
+
]]>
+ Python
+ Datamol
+ Pandas
+ Polars
+ itables
+ https://jhylin.github.io/Data_in_life_blog/posts/15_Molviz/itables.html
+ Wed, 09 Aug 2023 12:00:00 GMT
+
+
+ Working with scaffolds in small molecules
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/14_Scaffolds_in_small_molecules/chembl_anti-inf_data_prep_current.html
+
+
Features in post
+
This post will mainly be about the following:
+
+
Pre-process and standardise compounds (e.g. converting SMILES1 into SELFIES2 and other forms)
+
Obtain scaffolds for compounds
+
Align scaffolds of compounds
+
Query target scaffolds against a dataframe of compounds:
+
+
Function for saving multiple SMILES in .smi file
+
Function for converting .smi file into a list to query and match scaffolds of interests
+
Identify any similarities or differences in target compound of interest against other compounds in a dataframe
+
+
+
+
+
+
Quick words
+
I’ve always wanted to shorten my posts to a more readable length, but it was proven to be hard again, as this post was much longer than expected. Page content links are available on the right-hand side if needing to jump to sections for quick reads.
+
+
+
+
Key question to answer
+
Will the scaffold of compound 3 (compound of interest) be similar to the scaffolds of any approved anti-infectives in ChEMBL database?
+
+
+
+
Import libraries
+
The following libraries were used in this post.
+
+
import polars as pl
+import pandas as pd
+import datamol as dm
+import mols2grid
+
+from rdkit import Chem
+from rdkit.Chem import AllChem
+from rdkit.Chem.rdmolfiles import SmilesWriter, SmilesMolSupplier
+
+# Following library was modified & adapted from
+# Patrick Walters' tutorial on "Identifying Scaffolds"
+# - links provided in "scaffold_finder library" section under
+# the subheading of "Combining ChEMBL anti-infectives and FtsZ compounds"
+from scaffold_finder_test import find_scaffolds, get_molecules_with_scaffold
+
+
+
+
+
ChEMBL anti-infectives
+
+
Data cleaning
+
The dataset used was extracted from ChEMBL database, with a focus on the anti-infectives.
# Uncomment below if requiring a quick overview on all column names,
+# first ten variables in each column and each column data type
+#print(df_ai.glimpse())
+
+
Under the “Availability Type” column, there were a few different availabilities for each anti-bacterial such as, “Discontinued”, “Withdrawn”, “Unknown” and “Prescription Only”.
+
+
df_ai.groupby("Availability Type").count()
+
+
+
+
+shape: (4, 2)
+
+
+
+
+Availability Type
+
+
+count
+
+
+
+
+str
+
+
+u32
+
+
+
+
+
+
+"Discontinued"
+
+
+36
+
+
+
+
+"Withdrawn"
+
+
+7
+
+
+
+
+"Unknown"
+
+
+29
+
+
+
+
+"Prescription O...
+
+
+72
+
+
+
+
+
+
+
+
I only wanted to choose the “Prescription Only” ones, so the following filter condition was applied to the dataframe.
In preparation for possible future work on building machine learning models on this line of work, I looked into Datamol’s function on pre-processing molecules (shown in the next section), as it involved converting SMILES strings into SELFIES, which were considered to be more robust than SMILES.
+
However, I kept running into an error, with the error message showing the SMILES column was empty. After a few tries I realised that I’ve actually forgotten to check whether there were any missing SMILES in the column. So here I’ve filtered the SMILES column to look for any missing SMILES
+
+
df_ai_rx.filter(pl.col("Smiles") =="")
+
+
+
+
+shape: (1, 29)
+
+
+
+
+Parent Molecule
+
+
+Name
+
+
+Synonyms
+
+
+Research Codes
+
+
+Phase
+
+
+Drug Applicants
+
+
+USAN Stem
+
+
+USAN Year
+
+
+USAN Definition
+
+
+USAN Stem - Substem
+
+
+First Approval
+
+
+ATC Codes
+
+
+Level 4 ATC Codes
+
+
+Level 3 ATC Codes
+
+
+Level 2 ATC Codes
+
+
+Level 1 ATC Codes
+
+
+Indication Class
+
+
+Patent
+
+
+Drug Type
+
+
+Passes Rule of Five
+
+
+First In Class
+
+
+Chirality
+
+
+Prodrug
+
+
+Oral
+
+
+Parenteral
+
+
+Topical
+
+
+Black Box
+
+
+Availability Type
+
+
+Smiles
+
+
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+f64
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+i64
+
+
+i64
+
+
+str
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+str
+
+
+str
+
+
+
+
+
+
+"CHEMBL3989751"
+
+
+"NEOMYCIN"
+
+
+"FRADIOMYCIN|KA...
+
+
+""
+
+
+4.0
+
+
+"Bayer Pharmace...
+
+
+"'-mycin'"
+
+
+"1966"
+
+
+"antibiotics (S...
+
+
+"'-mycin(-mycin...
+
+
+"1957"
+
+
+"R02AB01 | S01A...
+
+
+"R02AB - Antibi...
+
+
+"R02A - THROAT ...
+
+
+"R02 - THROAT P...
+
+
+"R - RESPIRATOR...
+
+
+""
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+1
+
+
+0
+
+
+1
+
+
+1
+
+
+"Prescription O...
+
+
+""
+
+
+
+
+
+
+
+
Neomycin was the only compound found to have no SMILES recorded. To fix this error, I then used the “when-then-otherwise” expression in Polars again (used in previous post) to replace the empty string in the dataframe. A code example below was kindly adapted from StackOverflow from this link, and code example as shown below.
# Canonical SMILES for neomycin was extracted from PubChem
+# (https://pubchem.ncbi.nlm.nih.gov/compound/Neomycin)
+
+df_ai_rx = df_ai_rx.with_columns([
+ pl.when(pl.col("Smiles").str.lengths() ==0)
+ .then("C1C(C(C(C(C1N)OC2C(C(C(C(O2)CN)O)O)N)OC3C(C(C(O3)CO)OC4C(C(C(C(O4)CN)O)O)N)O)O)N")
+ .otherwise(pl.col("Smiles"))
+ .keep_name()
+])
+
+df_ai_rx
+
+
+
+
+shape: (72, 29)
+
+
+
+
+Parent Molecule
+
+
+Name
+
+
+Synonyms
+
+
+Research Codes
+
+
+Phase
+
+
+Drug Applicants
+
+
+USAN Stem
+
+
+USAN Year
+
+
+USAN Definition
+
+
+USAN Stem - Substem
+
+
+First Approval
+
+
+ATC Codes
+
+
+Level 4 ATC Codes
+
+
+Level 3 ATC Codes
+
+
+Level 2 ATC Codes
+
+
+Level 1 ATC Codes
+
+
+Indication Class
+
+
+Patent
+
+
+Drug Type
+
+
+Passes Rule of Five
+
+
+First In Class
+
+
+Chirality
+
+
+Prodrug
+
+
+Oral
+
+
+Parenteral
+
+
+Topical
+
+
+Black Box
+
+
+Availability Type
+
+
+Smiles
+
+
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+f64
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+i64
+
+
+i64
+
+
+str
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+str
+
+
+str
+
+
+
+
+
+
+"CHEMBL186"
+
+
+"CEFEPIME"
+
+
+"BMY-28142|CEFE...
+
+
+"BMY-28142|J01D...
+
+
+4.0
+
+
+"Samson Medical...
+
+
+"'cef-'"
+
+
+"1987"
+
+
+"cephalosporins...
+
+
+"'cef-(cef-)'"
+
+
+"1996"
+
+
+"J01DE01"
+
+
+"J01DE - Fourth...
+
+
+"J01D - OTHER B...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial"
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"CO/N=C(\C(=O)N...
+
+
+
+
+"CHEMBL2364632"
+
+
+"SARECYCLINE"
+
+
+"P-005672|P0056...
+
+
+"P-005672|P0056...
+
+
+4.0
+
+
+"Almirall Llc"
+
+
+"'-cycline'"
+
+
+"2012"
+
+
+"antibiotics (t...
+
+
+"'-cycline(-cyc...
+
+
+"2018"
+
+
+"J01AA14"
+
+
+"J01AA - Tetrac...
+
+
+"J01A - TETRACY...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+""
+
+
+"US-8318706-B2"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"CON(C)Cc1ccc(O...
+
+
+
+
+"CHEMBL31"
+
+
+"GATIFLOXACIN"
+
+
+"AM-1155|BMS-20...
+
+
+"AM-1155|BMS-20...
+
+
+4.0
+
+
+"Apotex Inc|Bri...
+
+
+"'-oxacin'"
+
+
+"1997"
+
+
+"antibacterials...
+
+
+"'-oxacin(-oxac...
+
+
+"1999"
+
+
+"S01AE06 | J01M...
+
+
+"S01AE - Fluoro...
+
+
+"S01A - ANTIINF...
+
+
+"S01 - OPHTHALM...
+
+
+"S - SENSORY OR...
+
+
+"Antibacterial"
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Racemic Mixtur...
+
+
+0
+
+
+1
+
+
+1
+
+
+1
+
+
+0
+
+
+"Prescription O...
+
+
+"COc1c(N2CCNC(C...
+
+
+
+
+"CHEMBL3039597"
+
+
+"GENTAMICIN"
+
+
+"GENTAMICIN|GEN...
+
+
+"SCH-9724"
+
+
+4.0
+
+
+"Schering Corp ...
+
+
+"'-micin'"
+
+
+"1963"
+
+
+"antibiotics (M...
+
+
+"'-micin(-micin...
+
+
+"1970"
+
+
+"S01AA11 | S02A...
+
+
+"S01AA - Antibi...
+
+
+"S01A - ANTIINF...
+
+
+"S01 - OPHTHALM...
+
+
+"S - SENSORY OR...
+
+
+"Antibacterial"
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Unknown"
+
+
+0
+
+
+0
+
+
+1
+
+
+1
+
+
+1
+
+
+"Prescription O...
+
+
+"CNC(C)[C@@H]1C...
+
+
+
+
+"CHEMBL893"
+
+
+"DICLOXACILLIN"
+
+
+"BRL-1702|DICLO...
+
+
+"BRL-1702|R-134...
+
+
+4.0
+
+
+"Teva Pharmaceu...
+
+
+"'-cillin'"
+
+
+"1965"
+
+
+"penicillins"
+
+
+"'-cillin(-cill...
+
+
+"1968"
+
+
+"J01CF01"
+
+
+"J01CF - Beta-l...
+
+
+"J01C - BETA-LA...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial"
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"Cc1onc(-c2c(Cl...
+
+
+
+
+"CHEMBL1449"
+
+
+"TICARCILLIN"
+
+
+"TICARCILLIN|Ti...
+
+
+""
+
+
+4.0
+
+
+"Glaxosmithklin...
+
+
+"'-cillin'"
+
+
+"1973"
+
+
+"penicillins"
+
+
+"'-cillin(-cill...
+
+
+"1976"
+
+
+"J01CA13"
+
+
+"J01CA - Penici...
+
+
+"J01C - BETA-LA...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial"
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"CC1(C)S[C@@H]2...
+
+
+
+
+"CHEMBL1220"
+
+
+"TINIDAZOLE"
+
+
+"CP-12,574|CP-1...
+
+
+"CP-12,574|CP-1...
+
+
+4.0
+
+
+"Mission Pharma...
+
+
+"'-nidazole'"
+
+
+"1970"
+
+
+"antiprotozoal ...
+
+
+"'-nidazole(-ni...
+
+
+"2004"
+
+
+"P01AB02 | J01X...
+
+
+"P01AB - Nitroi...
+
+
+"P01A - AGENTS ...
+
+
+"P01 - ANTIPROT...
+
+
+"P - ANTIPARASI...
+
+
+"Antiprotozoal"
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Achiral Molecu...
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+1
+
+
+"Prescription O...
+
+
+"CCS(=O)(=O)CCn...
+
+
+
+
+"CHEMBL501122"
+
+
+"CEFTAROLINE FO...
+
+
+"CEFTAROLINE FO...
+
+
+"PPI-0903|TAK 5...
+
+
+4.0
+
+
+"Apotex Inc|All...
+
+
+"'cef-; fos-'"
+
+
+"2006"
+
+
+"cephalosporins...
+
+
+"'cef-(cef-); f...
+
+
+"2010"
+
+
+"J01DI02"
+
+
+"J01DI - Other ...
+
+
+"J01D - OTHER B...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+""
+
+
+"US-6417175-B1"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Single Stereoi...
+
+
+1
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"CCO/N=C(\C(=O)...
+
+
+
+
+"CHEMBL137"
+
+
+"METRONIDAZOLE"
+
+
+"ACEA|ANABACT|B...
+
+
+"BAY-5360|BAYER...
+
+
+4.0
+
+
+"Inforlife Sa|L...
+
+
+"'-nidazole'"
+
+
+"1962"
+
+
+"antiprotozoal ...
+
+
+"'-nidazole(-ni...
+
+
+"1963"
+
+
+"D06BX01 | P01A...
+
+
+"D06BX - Other ...
+
+
+"D06B - CHEMOTH...
+
+
+"D06 - ANTIBIOT...
+
+
+"D - DERMATOLOG...
+
+
+"Antibacterial,...
+
+
+"US-6881726-B2"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Achiral Molecu...
+
+
+0
+
+
+1
+
+
+1
+
+
+1
+
+
+1
+
+
+"Prescription O...
+
+
+"Cc1ncc([N+](=O...
+
+
+
+
+"CHEMBL376140"
+
+
+"TIGECYCLINE"
+
+
+"TIGECYCLINE|Ty...
+
+
+"WAY-GAR-936"
+
+
+4.0
+
+
+"Apotex Inc|Fre...
+
+
+"'-cycline'"
+
+
+"2002"
+
+
+"antibiotics (t...
+
+
+"'-cycline(-cyc...
+
+
+"2005"
+
+
+"J01AA12"
+
+
+"J01AA - Tetrac...
+
+
+"J01A - TETRACY...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+""
+
+
+"US-7879828-B2"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+0
+
+
+1
+
+
+0
+
+
+1
+
+
+"Prescription O...
+
+
+"CN(C)c1cc(NC(=...
+
+
+
+
+"CHEMBL1741"
+
+
+"CLARITHROMYCIN...
+
+
+"6-O-METHYLERYT...
+
+
+"A-56268|ABBOTT...
+
+
+4.0
+
+
+"Sunshine Lake ...
+
+
+"'-mycin'"
+
+
+"1988"
+
+
+"antibiotics (S...
+
+
+"'-mycin(-mycin...
+
+
+"1991"
+
+
+"J01FA09"
+
+
+"J01FA - Macrol...
+
+
+"J01F - MACROLI...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial"
+
+
+"US-7977488-B2"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"CC[C@H]1OC(=O)...
+
+
+
+
+"CHEMBL1747"
+
+
+"TOBRAMYCIN"
+
+
+"47663|Aktob|BE...
+
+
+"47663|NSC-1805...
+
+
+4.0
+
+
+"Igi Laboratori...
+
+
+"'-mycin'"
+
+
+"1972"
+
+
+"antibiotics (S...
+
+
+"'-mycin(-mycin...
+
+
+"1975"
+
+
+"J01GB01 | S01A...
+
+
+"J01GB - Other ...
+
+
+"J01G - AMINOGL...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial"
+
+
+"US-6987094-B2"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+0
+
+
+1
+
+
+1
+
+
+1
+
+
+"Prescription O...
+
+
+"NC[C@H]1O[C@H]...
+
+
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+...
+
+
+
+
+"CHEMBL404"
+
+
+"TAZOBACTAM"
+
+
+"CL 298,741|CL-...
+
+
+"CL 298,741|CL-...
+
+
+4.0
+
+
+"Wytells Pharma...
+
+
+"'-bactam'"
+
+
+"1989"
+
+
+"beta-lactamase...
+
+
+"'-bactam(-bact...
+
+
+"1993"
+
+
+"J01CG02"
+
+
+"J01CG - Beta-l...
+
+
+"J01C - BETA-LA...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Inhibitor (bet...
+
+
+"US-6900184-B2"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"C[C@]1(Cn2ccnn...
+
+
+
+
+"CHEMBL1689772"
+
+
+"OMADACYCLINE"
+
+
+"AMADACYCLINE|B...
+
+
+"BAY 73-6944|PT...
+
+
+4.0
+
+
+"Paratek Pharma...
+
+
+"'-cycline'"
+
+
+"2009"
+
+
+"antibiotics (t...
+
+
+"'-cycline(-cyc...
+
+
+"2018"
+
+
+"J01AA15"
+
+
+"J01AA - Tetrac...
+
+
+"J01A - TETRACY...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+""
+
+
+"US-7326696-B2"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"CN(C)c1cc(CNCC...
+
+
+
+
+"CHEMBL3989974"
+
+
+"CEFIDEROCOL"
+
+
+"CEFIDEROCOL|GS...
+
+
+"GSK2696266|S-6...
+
+
+4.0
+
+
+"Shionogi Inc"
+
+
+"'cef-'"
+
+
+"2017"
+
+
+"cephalosporins...
+
+
+"'cef-(cef-)'"
+
+
+"2019"
+
+
+"J01DI04"
+
+
+"J01DI - Other ...
+
+
+"J01D - OTHER B...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+""
+
+
+"US-9238657-B2"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"CC(C)(O/N=C(\C...
+
+
+
+
+"CHEMBL1435"
+
+
+"CEFAZOLIN"
+
+
+"CEFAZOLIN|CEPH...
+
+
+"J01DB04|SK&F-4...
+
+
+4.0
+
+
+"Glaxosmithklin...
+
+
+"'cef-'"
+
+
+"1972"
+
+
+"cephalosporins...
+
+
+"'cef-(cef-)'"
+
+
+"1973"
+
+
+"J01DB04"
+
+
+"J01DB - First-...
+
+
+"J01D - OTHER B...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial ...
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"Cc1nnc(SCC2=C(...
+
+
+
+
+"CHEMBL3989751"
+
+
+"NEOMYCIN"
+
+
+"FRADIOMYCIN|KA...
+
+
+""
+
+
+4.0
+
+
+"Bayer Pharmace...
+
+
+"'-mycin'"
+
+
+"1966"
+
+
+"antibiotics (S...
+
+
+"'-mycin(-mycin...
+
+
+"1957"
+
+
+"R02AB01 | S01A...
+
+
+"R02AB - Antibi...
+
+
+"R02A - THROAT ...
+
+
+"R02 - THROAT P...
+
+
+"R - RESPIRATOR...
+
+
+""
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+1
+
+
+0
+
+
+1
+
+
+1
+
+
+"Prescription O...
+
+
+"C1C(C(C(C(C1N)...
+
+
+
+
+"CHEMBL572"
+
+
+"NITROFURANTOIN...
+
+
+"BERKFURIN|CEDU...
+
+
+"NSC-2107|NSC-4...
+
+
+4.0
+
+
+"Sun Pharmaceut...
+
+
+"'-toin'"
+
+
+""
+
+
+"antiepileptics...
+
+
+"'-toin(-toin)'...
+
+
+"1953"
+
+
+"J01XE01 | J01X...
+
+
+"J01XE - Nitrof...
+
+
+"J01X - OTHER A...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial ...
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Achiral Molecu...
+
+
+0
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+"Prescription O...
+
+
+"O=C1CN(/N=C/c2...
+
+
+
+
+"CHEMBL44354"
+
+
+"CEFTAZIDIME"
+
+
+"CEFTAZIDIME|CE...
+
+
+"GR 20263|GR-20...
+
+
+4.0
+
+
+"Glaxosmithklin...
+
+
+"'cef-'"
+
+
+"1980"
+
+
+"cephalosporins...
+
+
+"'cef-(cef-)'"
+
+
+"1985"
+
+
+"J01DD02"
+
+
+"J01DD - Third-...
+
+
+"J01D - OTHER B...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial"
+
+
+"US-7112592-B2"
+
+
+"1:Synthetic Sm...
+
+
+0
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+0
+
+
+1
+
+
+0
+
+
+1
+
+
+"Prescription O...
+
+
+"CC(C)(O/N=C(\C...
+
+
+
+
+"CHEMBL29"
+
+
+"BENZYLPENICILL...
+
+
+"BENZYL PENICIL...
+
+
+"J01CE01|NSC-19...
+
+
+4.0
+
+
+"Hq Specialty P...
+
+
+"'-cillin'"
+
+
+""
+
+
+"penicillins"
+
+
+"'-cillin(-cill...
+
+
+"1947"
+
+
+"J01CE01 | S01A...
+
+
+"J01CE - Beta-l...
+
+
+"J01C - BETA-LA...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial"
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Single Stereoi...
+
+
+0
+
+
+1
+
+
+1
+
+
+0
+
+
+1
+
+
+"Prescription O...
+
+
+"CC1(C)S[C@@H]2...
+
+
+
+
+"CHEMBL8"
+
+
+"CIPROFLOXACIN"
+
+
+"BAY O 9867 FRE...
+
+
+"BAY O 9867 FRE...
+
+
+4.0
+
+
+"Inforlife Sa|A...
+
+
+"'-oxacin'"
+
+
+"1987"
+
+
+"antibacterials...
+
+
+"'-oxacin(-oxac...
+
+
+"1987"
+
+
+"J01MA02 | S03A...
+
+
+"J01MA - Fluoro...
+
+
+"J01M - QUINOLO...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial"
+
+
+"US-8318817-B2"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Achiral Molecu...
+
+
+0
+
+
+1
+
+
+1
+
+
+1
+
+
+1
+
+
+"Prescription O...
+
+
+"O=C(O)c1cn(C2C...
+
+
+
+
+"CHEMBL9"
+
+
+"NORFLOXACIN"
+
+
+"Baccidal|CHIBR...
+
+
+"MK-366|NSC-757...
+
+
+4.0
+
+
+"Merck Research...
+
+
+"'-oxacin'"
+
+
+"1984"
+
+
+"antibacterials...
+
+
+"'-oxacin(-oxac...
+
+
+"1986"
+
+
+"J01MA06 | S01A...
+
+
+"J01MA - Fluoro...
+
+
+"J01M - QUINOLO...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial"
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Achiral Molecu...
+
+
+0
+
+
+1
+
+
+0
+
+
+1
+
+
+1
+
+
+"Prescription O...
+
+
+"CCn1cc(C(=O)O)...
+
+
+
+
+"CHEMBL21"
+
+
+"SULFANILAMIDE"
+
+
+"ANILINE-P-SULF...
+
+
+"NSC-7618"
+
+
+4.0
+
+
+"Mylan Specialt...
+
+
+"'sulfa-'"
+
+
+""
+
+
+"antimicrobials...
+
+
+"'sulfa-(sulfa-...
+
+
+"1985"
+
+
+"D06BA05 | J01E...
+
+
+"D06BA - Sulfon...
+
+
+"D06B - CHEMOTH...
+
+
+"D06 - ANTIBIOT...
+
+
+"D - DERMATOLOG...
+
+
+""
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Achiral Molecu...
+
+
+0
+
+
+0
+
+
+0
+
+
+1
+
+
+0
+
+
+"Prescription O...
+
+
+"Nc1ccc(S(N)(=O...
+
+
+
+
+"CHEMBL4"
+
+
+"OFLOXACIN"
+
+
+"DL-8280|EXOCIN...
+
+
+"DL-8280|HOE 28...
+
+
+4.0
+
+
+"Bausch And Lom...
+
+
+"'-oxacin'"
+
+
+"1984"
+
+
+"antibacterials...
+
+
+"'-oxacin(-oxac...
+
+
+"1990"
+
+
+"J01MA01 | S02A...
+
+
+"J01MA - Fluoro...
+
+
+"J01M - QUINOLO...
+
+
+"J01 - ANTIBACT...
+
+
+"J - ANTIINFECT...
+
+
+"Antibacterial"
+
+
+"None"
+
+
+"1:Synthetic Sm...
+
+
+1
+
+
+0
+
+
+"Racemic Mixtur...
+
+
+0
+
+
+1
+
+
+1
+
+
+1
+
+
+1
+
+
+"Prescription O...
+
+
+"CC1COc2c(N3CCN...
+
+
+
+
+
+
+
+
+
# Keeping only selected columns with information needed for later use
+df_ai_rx = df_ai_rx.select(["Smiles", "Name", "USAN Definition", "Level 4 ATC Codes"])
+df_ai_rx.head()
+
+
+
+
+shape: (5, 4)
+
+
+
+
+Smiles
+
+
+Name
+
+
+USAN Definition
+
+
+Level 4 ATC Codes
+
+
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+
+
+
+
+"CO/N=C(\C(=O)N...
+
+
+"CEFEPIME"
+
+
+"cephalosporins...
+
+
+"J01DE - Fourth...
+
+
+
+
+"CON(C)Cc1ccc(O...
+
+
+"SARECYCLINE"
+
+
+"antibiotics (t...
+
+
+"J01AA - Tetrac...
+
+
+
+
+"COc1c(N2CCNC(C...
+
+
+"GATIFLOXACIN"
+
+
+"antibacterials...
+
+
+"S01AE - Fluoro...
+
+
+
+
+"CNC(C)[C@@H]1C...
+
+
+"GENTAMICIN"
+
+
+"antibiotics (M...
+
+
+"S01AA - Antibi...
+
+
+
+
+"Cc1onc(-c2c(Cl...
+
+
+"DICLOXACILLIN"
+
+
+"penicillins"
+
+
+"J01CF - Beta-l...
+
+
+
+
+
+
+
+
The “Smiles” column name was changed below to ensure _preprocess function would work since the parameter “smiles_column” in _preprocess function had “smiles” with lowercase “s” (this of course could be the other way round, where we could change the parameter name in the function instead - the column name and parameter name had to match for the function to work). The “Name” column was changed accordingly for similar reason.
I also wanted to change the all capitalised compound names into lowercases for the ease of reading.
+
+
# Convert all compounds to lowercases
+df_ai_rx = df_ai_rx.with_columns(pl.col("names").str.to_lowercase())
+df_ai_rx.head()
+
+
+
+
+shape: (5, 4)
+
+
+
+
+smiles
+
+
+names
+
+
+USAN Definition
+
+
+Level 4 ATC Codes
+
+
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+
+
+
+
+"CO/N=C(\C(=O)N...
+
+
+"cefepime"
+
+
+"cephalosporins...
+
+
+"J01DE - Fourth...
+
+
+
+
+"CON(C)Cc1ccc(O...
+
+
+"sarecycline"
+
+
+"antibiotics (t...
+
+
+"J01AA - Tetrac...
+
+
+
+
+"COc1c(N2CCNC(C...
+
+
+"gatifloxacin"
+
+
+"antibacterials...
+
+
+"S01AE - Fluoro...
+
+
+
+
+"CNC(C)[C@@H]1C...
+
+
+"gentamicin"
+
+
+"antibiotics (M...
+
+
+"S01AA - Antibi...
+
+
+
+
+"Cc1onc(-c2c(Cl...
+
+
+"dicloxacillin"
+
+
+"penicillins"
+
+
+"J01CF - Beta-l...
+
+
+
+
+
+
+
+
Since Datamol was built as a thin layer library on top of RDKit, which was really only compatible with Pandas, I added the following step to convert the dataframe into a Pandas one.
+
+
df_ai_pd = df_ai_rx.to_pandas()
+df_ai_pd.head()
+
+
+
+
+
+
+
+
+
smiles
+
names
+
USAN Definition
+
Level 4 ATC Codes
+
+
+
+
+
0
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
cefepime
+
cephalosporins
+
J01DE - Fourth-generation cephalosporins
+
+
+
1
+
CON(C)Cc1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C...
+
sarecycline
+
antibiotics (tetracycline derivatives)
+
J01AA - Tetracyclines
+
+
+
2
+
COc1c(N2CCNC(C)C2)c(F)cc2c(=O)c(C(=O)O)cn(C3CC...
+
gatifloxacin
+
antibacterials (quinolone derivatives)
+
S01AE - Fluoroquinolones | J01MA - Fluoroquino...
+
+
+
3
+
CNC(C)[C@@H]1CC[C@@H](N)[C@@H](O[C@H]2[C@H](O)...
+
gentamicin
+
antibiotics (Micromonospora strains)
+
S01AA - Antibiotics | S02AA - Antiinfectives |...
+
+
+
4
+
Cc1onc(-c2c(Cl)cccc2Cl)c1C(=O)N[C@@H]1C(=O)N2[...
+
dicloxacillin
+
penicillins
+
J01CF - Beta-lactamase resistant penicillins
+
+
+
+
+
+
+
+
# Check the dataframe has been converted from Polars to Pandas
+type(df_ai_pd)
+
+
pandas.core.frame.DataFrame
+
+
+
+
+
+
Pre-processing and standardising molecules
+
I have borrowed and adapted the _preprocess function from Datamol (link here), as shown below. One of the convenient features in this function was that it also included a conversion from “mol” (RDKit molecule) to SELFIES amongst several other common molecular representations such as InChI3 and SMILES.
The images generated below might be quite small to see or read clearly. I’ve tried to increase the molecule size (mol_size) and also reduce the column numbers, but it still appeared the same. However, if the code was run in say VS Code, the compound images would appear larger when increasing the mol_size.
+
+
# Grab all SMILES of the cleaned/pre-processed ChEMBL anti-infectives
+df_ai_sm = data_mol_clean["standard_smiles"]
+
+# Load a list of these molecules in SMILES
+# dm.to_mol() has sanitize = True set as default
+mol_ls = [dm.to_mol(smile) for smile in df_ai_sm]
+
+# Alternative way to convert dataframe into a list of mols (same as mol_ls)
+# mols = dm.from_df(df_name, smiles_column = "Smiles")
+
+# Add compound name for each 2D image
+legends_c =list(data_mol_clean["names"])
+
+# Convert the list of molecules into 2D images
+dm.to_image(mol_ls, n_cols =4, mol_size = (400, 400), legends = legends_c)
+
+
+
+
+
+
+
+
Extract scaffolds
+
+
# Extract Murcko scaffolds from mol_ls (ChEMBL anti-infectives)
+m_scaffolds = [dm.to_scaffold_murcko(mol) for mol in mol_ls]
+dm.to_image(m_scaffolds, mol_size = (400, 400), legends = legends_c)
+
+
+
+
+
+
+
+
+
Filamenting temperature-sensitive mutant Z (FtsZ) compounds
Before I started cleaning any data on FtsZ compounds, I found this useful website, OPSIN: Open Parser for Systematic IUPAC nomenclature, with this link to the journal paper as an acknowledgement of the work. I’ve managed to convert these 3 FtsZ compounds by using their IUPAC names, which were inputted into OPSIN, and converted into the corresponding InChI or SMILES strings.
+
After that, I started by converting the InChI of compound 1 into a RDKit molecule, which could be visualised in 2D below.
+
+
# Convert compound 1 to mol from InChI
+cpd1 = dm.from_inchi("InChI=1S/C22H20O4/c23-18-9-4-15(5-10-18)8-13-21(25)20-3-1-2-17(22(20)26)14-16-6-11-19(24)12-7-16/h4-14,20,23-24H,1-3H2/b13-8+,17-14+")
+cpd1
+
+
+
+
+
I then converted compound 2 using SMILES string instead.
# Grab all SMILES from cleaned FtsZ compound dataset
+df_ai_ftsz = data_cleaned["standard_smiles"]
+
+# Load a list of these molecules in SMILES
+mol_ftsz_list = [dm.to_mol(smile) for smile in df_ai_ftsz]
+
+# Add compound names for each 2D image of compounds
+legends =list(data_cleaned["names"])
+
+# Convert the list of molecules into 2D images
+dm.to_image(mol_ftsz_list, n_cols =5, mol_size = (400, 400), legends = legends)
+
+
+
+
+
+
+
+
Extract scaffolds
+
+
# Get Murcko scaffolds of FtsZ compounds
+m_ftsz_scaffolds = [dm.to_scaffold_murcko(mol) for mol in mol_ftsz_list]
+dm.to_image(m_ftsz_scaffolds, mol_size = (400, 400), legends = legends)
+
+
+
+
+
+
+
+
+
Combining ChEMBL anti-infectives and FtsZ compounds
+
+
Combining dataframes
+
In this part, I wanted to combine the two dataframes I had from above, since my next step was to compare the scaffolds between ChEMBL prescription-only anti-infectives and FtsZ compounds.
# Convert the standard SMILES into RDKit molecules
+mol_full = [dm.to_mol(smile) for smile in df_full]
+
+
+
+
+
Aligning all the scaffolds
+
Here, all the scaffolds from both dataframes of compounds were aligned by using Datamol’s auto_align_many(). The images of all the aligned molecules were generated in the end. The compound structures did re-align, but unfortunately it only showed up to a maximum of 50 compounds only (the maximum number of molecules to be shown in Datamol was set to 32 as default; this number was pushed up to and truncated at 50 in the warning message from RDKit when attempting to run a total of 75 compounds using the Datamol library, without looking further into other ways to alter this for now).
An attempt to combine Datamol’s auto_align_many() with mols2grid library was shown below. Unfortunately, the compounds did not re-align but all 75 compounds were shown in the grids.
+
+
mols2grid.display(aligned_list)
+
+
+
+
+
+
+
+
+
+
mols2grid library
+
Since I’ve started using mols2grid here, I thought to show an example of this library by combining all 75 compounds using the pre-processed standard SMILES in the grids with corresponding compound names. The resulting table provided a clear overview of all the compounds, with useful options to select or filter compounds. Obviously, other molecular properties or experimental results could be added into the table for other uses.
+
+
# Full dataset of 75 compounds
+mols2grid.display(full_data, smiles_col ="standard_smiles", subset = ["img", "mols2grid-id", "names"])
+
+
+
+
+
+
+
+
+
+
+
scaffold_finder library
+
Rather than only trying out Datamol only, I also thought to try out the scaffold_finder library after reading this Jupyter notebook by Patrick Walters. The GitHub repository of his other useful cheminformatics tutorials can be found here. His blog is here. Without surprises, this post was also inspired by this Jupyter notebook on “Identifying scaffolds in a set of molecules”, with some hope to expand on it a bit more.
+
Below were my notes on how to use this particular library.
+
Step 1: Add “mol” column to full_data dataframe (this was needed in order to use the functions from scaffold_finder library, which was also built based on RDKit)
Step 2: Change column names of “standard_smiles” to “SMILES” & “names” to “Name” to match with scaffold_finder library functions with set column names (or other way round, by changing the names in the function library)
+
+
# Note: New column name "SMILES" contains standardised SMILES (old column name as "standard_smiles")
+full_data = full_data.rename(columns = {"standard_smiles": "SMILES", "names": "Name"})
+full_data.head()
+
+
+
+
+
+
+
+
+
smiles
+
Name
+
USAN Definition
+
Level 4 ATC Codes
+
SMILES
+
selfies
+
inchi
+
inchikey
+
mol
+
+
+
+
+
0
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
cefepime
+
cephalosporins
+
J01DE - Fourth-generation cephalosporins
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
[C][O][/N][=C][Branch2][Ring2][#Branch2][\C][=...
+
InChI=1S/C19H24N6O5S2/c1-25(5-3-4-6-25)7-10-8-...
+
HVFLCNVBZFFHBT-ZKDACBOMSA-N
+
<rdkit.Chem.rdchem.Mol object at 0x134307680>
+
+
+
1
+
CON(C)Cc1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C...
+
sarecycline
+
antibiotics (tetracycline derivatives)
+
J01AA - Tetracyclines
+
CON(C)Cc1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C...
+
[C][O][N][Branch1][C][C][C][C][=C][C][=C][Bran...
+
InChI=1S/C24H29N3O8/c1-26(2)18-13-8-11-7-12-10...
+
PQJQFLNBMSCUSH-SBAJWEJLSA-N
+
<rdkit.Chem.rdchem.Mol object at 0x134306c00>
+
+
+
2
+
COc1c(N2CCNC(C)C2)c(F)cc2c(=O)c(C(=O)O)cn(C3CC...
+
gatifloxacin
+
antibacterials (quinolone derivatives)
+
S01AE - Fluoroquinolones | J01MA - Fluoroquino...
+
COc1c(N2CCNC(C)C2)c(F)cc2c(=O)c(C(=O)O)cn(C3CC...
+
[C][O][C][=C][Branch1][N][N][C][C][N][C][Branc...
+
InChI=1S/C19H22FN3O4/c1-10-8-22(6-5-21-10)16-1...
+
XUBOMFCQGDBHNK-UHFFFAOYSA-N
+
<rdkit.Chem.rdchem.Mol object at 0x133e729d0>
+
+
+
3
+
CNC(C)[C@@H]1CC[C@@H](N)[C@@H](O[C@H]2[C@H](O)...
+
gentamicin
+
antibiotics (Micromonospora strains)
+
S01AA - Antibiotics | S02AA - Antiinfectives |...
+
CNC(C)[C@@H]1CC[C@@H](N)[C@@H](O[C@H]2[C@H](O)...
+
[C][N][C][Branch1][C][C][C@@H1][C][C][C@@H1][B...
+
InChI=1S/C21H43N5O7.C20H41N5O7.C19H39N5O7/c1-9...
+
NPEFREDMMVQEPL-RWPARATISA-N
+
<rdkit.Chem.rdchem.Mol object at 0x133e72500>
+
+
+
4
+
Cc1onc(-c2c(Cl)cccc2Cl)c1C(=O)N[C@@H]1C(=O)N2[...
+
dicloxacillin
+
penicillins
+
J01CF - Beta-lactamase resistant penicillins
+
Cc1onc(-c2c(Cl)cccc2Cl)c1C(=O)N[C@@H]1C(=O)N2[...
+
[C][C][O][N][=C][Branch1][=N][C][=C][Branch1][...
+
InChI=1S/C19H17Cl2N3O5S/c1-7-10(12(23-29-7)11-...
+
YFAGHNZHGGCZAX-JKIFEVAISA-N
+
<rdkit.Chem.rdchem.Mol object at 0x133e719a0>
+
+
+
+
+
+
+
Step 3: Identify scaffolds
+
The find_scaffolds() function was kindly borrowed from the scaffold_finder library as mentioned above. The scaffold_finder_test.py was the modified version, as I’ve used a different dataset here.
+
+
mol_df, scaffold_df = find_scaffolds(full_data)
+
+
+
+
+
Below was a quick overview of the mol_df, showing scaffolds in SMILES, number of atoms, number of R groups, names of compounds and the standardised SMILES of the compounds.
+
+
mol_df
+
+
+
+
+
+
+
+
+
Scaffold
+
NumAtoms
+
NumRgroupgs
+
Name
+
SMILES
+
+
+
+
+
0
+
C[N+]1(CC2=C(C(=O)[O-])N3C(=O)[C@@H](NC(=O)/C(...
+
31
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
1
+
C[N+]1(CC2=CN3C(=O)[C@@H](NC(=O)C(=NO)c4csc(N)...
+
28
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
2
+
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=CCS[C@H]23)cs1
+
21
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
3
+
CC1=CN2C(=O)[C@@H](NC(=O)C(=NO)c3csc(N)n3)[C@H...
+
22
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
4
+
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=C(C[NH+]4CCC...
+
27
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
...
+
...
+
...
+
...
+
...
+
...
+
+
+
15
+
O=C(CCc1ccccc1)C1CCCC(Cc2ccccc2)C1=O
+
24
+
2
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
16
+
O=C1CCCCC1C(=O)CCc1ccc(O)cc1
+
18
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
19
+
CC1CCCC(C(=O)CCc2ccc(O)cc2)C1=O
+
19
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
21
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccccc2)C1=O
+
25
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
22
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
26
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
+
5320 rows × 5 columns
+
+
+
+
Again to have a quick look at the scaffolds of all 75 compounds, along with counts of each scaffold and number of atoms in each scaffold.
+
+
scaffold_df
+
+
+
+
+
+
+
+
+
Scaffold
+
Count
+
NumAtoms
+
+
+
+
+
1156
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
+
7
+
29
+
+
+
1101
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
6
+
30
+
+
+
173
+
CC(=O)N[C@@H]1C(=O)N2[C@@H]1SC(C)(C)[C@@H]2C(=O)O
+
5
+
17
+
+
+
174
+
CC(=O)N[C@@H]1C(=O)N2[C@@H]1SC(C)[C@@H]2C(=O)O
+
5
+
16
+
+
+
552
+
CC1(C)S[C@@H]2[C@H](NC=O)C(=O)N2[C@H]1C(=O)O
+
5
+
16
+
+
+
...
+
...
+
...
+
...
+
+
+
3684
+
Cc1nccn1C
+
1
+
7
+
+
+
4456
+
Nc1ccccc1
+
1
+
7
+
+
+
4775
+
O=P(O)(O)[C@@H]1CO1
+
1
+
7
+
+
+
3722
+
Cn1ccnc1
+
1
+
6
+
+
+
4807
+
c1ccccc1
+
1
+
6
+
+
+
+
4808 rows × 3 columns
+
+
+
+
Step 4: Display all scaffolds in mols2grid, which helped to identify the scaffold with the highest frequency (counts) of occurence in the dataset.
These were my sample datasets for later use in the section on “Reading and querying multiple scaffolds in SMILES strings”.
+
Below was the first test dataset on the top 2 scaffolds with highest frequency of appearance in the full dataframe.
+
+
# Scaffold of anti-infective with highest count
+count_top1_scaffold = scaffold_df.Scaffold.values[0]
+# Scaffold of anti-infective with the second highest count
+count_top2_scaffold = scaffold_df.Scaffold.values[1]
+
+
+
# Combine above scaffolds into a list
+count_top_scaffold =list((count_top1_scaffold, count_top2_scaffold))
+count_top_scaffold
Compound 3 was the compound found in the paper to have targeted the FtsZ proteins in Gram positive pathogens such as Streptococcus pneumoniae with more pronounced activities than its predecessor e.g. compound 1. So this section aimed to look into all of compound 3’s scaffolds.
+
+
# For ease of dataframe manipulation, decided to convert Pandas df into a Polars one (just my personal preference as I've used Polars more lately)
+# then filtered out all the scaffolds for compound 3 & saved it as an independent dataframe
+cpd3_scaffolds = pl.from_pandas(mol_df).filter(pl.col("Name") =="Compound_3")
+cpd3_scaffolds
+
+
+
+
+shape: (14, 5)
+
+
+
+
+Scaffold
+
+
+NumAtoms
+
+
+NumRgroupgs
+
+
+Name
+
+
+SMILES
+
+
+
+
+str
+
+
+i64
+
+
+i64
+
+
+str
+
+
+str
+
+
+
+
+
+
+"O=CC1CCCC(Cc2c...
+
+
+17
+
+
+1
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"O=CC1CCCC(Cc2c...
+
+
+16
+
+
+2
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"CC(=O)C1CCCC(C...
+
+
+18
+
+
+1
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"CC(=O)C1CCCC(C...
+
+
+17
+
+
+2
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"CCC(=O)C1CCCC(...
+
+
+19
+
+
+1
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"CCC(=O)C1CCCC(...
+
+
+18
+
+
+2
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"O=C(CCc1ccccc1...
+
+
+25
+
+
+1
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"O=C1CCCCC1C(=O...
+
+
+17
+
+
+2
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"CC1CCCC(C(=O)C...
+
+
+18
+
+
+2
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"O=C(CCc1ccccc1...
+
+
+24
+
+
+2
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"O=C1CCCCC1C(=O...
+
+
+18
+
+
+1
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"CC1CCCC(C(=O)C...
+
+
+19
+
+
+1
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"O=C(CCc1ccc(O)...
+
+
+25
+
+
+1
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+"O=C(CCc1ccc(O)...
+
+
+26
+
+
+1
+
+
+"Compound_3"
+
+
+"O=C(CCc1ccc(O)...
+
+
+
+
+
+
+
+
+
# Convert Polars df into a Pandas one
+# and use mols2grid to show the 2D images of compound 3 scaffolds
+# Total of 14 different scaffolds
+cpd3_scaffolds = cpd3_scaffolds.to_pandas()
+mols2grid.display(cpd3_scaffolds, smiles_col ="Scaffold")
+
+
+
+
+
+
+
+
+
Testing compound 3 scaffolds using scaffold_finder library
+
At this stage, I sort of had an idea of wanting to compare all 14 compound 3 scaffolds against all 75 molecules including ChEMBL-curated prescription-only anti-bacterials.
+
I tried the get_molecules_with_scaffold() function from scaffold_finder library but didn’t exactly get what I hoped to achieve. I played around a bit and noticed it was really designed for spotting a single target scaffold with highest counts in the data set. I was hoping to parse multiple scaffolds actually, or imagining there might be situations where we might want to do this.
+
I started trialling with one scaffold anyway as shown below on the get_molecule_with_scaffold() function from the scaffold_finder library.
+
+
# Trial single scaffold first
+scaffold_test = cpd3_scaffolds.Scaffold.values[0]
+scaffold_test
# Showing only compound 3 as a distinctive compound (no other molecules with similar scaffold)
+chem_mol_df
+
+
+
+
+
+
+
+
+
SMILES
+
Name
+
+
+
+
+
0
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
Compound_3
+
+
+
+
+
+
+
+
+
+
+
Reading and querying multiple scaffolds in SMILES strings
+
I also tried to tweak the get_molecule_with_scaffolds() function but realised it might be even better to write my own function to tailor to my need. Therefore, I wrote a small and simple function which would read and query multiple scaffolds of small molecules in SMILES string formats against a dataframe (with information showing scaffolds in SMILES, number of atoms, number of R groups, names of compounds and also the SMILES of the compounds).
+
At first, I started with reading all 14 scaffolds of compound 3 by using the values index method on cpd3_scaffolds dataframe that included all 14 scaffolds of compound 3.
+
+
# Trial feeding all 14 SMILES
+scaffold_cpd3_all = cpd3_scaffolds.Scaffold.values[:]
+scaffold_cpd3_all
Then I thought about how every time if we’d want to convert any molecules from SMILES to RDKit molecules, we really had to have a “mol” column set up, so that was what I did below.
Then perhaps I would place all of these compound 3 scaffolds into an object.
+
+
cpd3_mols = cpd3_scaffolds["mol"]
+cpd3_mols
+
+
0 <rdkit.Chem.rdchem.Mol object at 0x13580d3f0>
+1 <rdkit.Chem.rdchem.Mol object at 0x13580d4d0>
+2 <rdkit.Chem.rdchem.Mol object at 0x13580dd20>
+3 <rdkit.Chem.rdchem.Mol object at 0x13580e880>
+4 <rdkit.Chem.rdchem.Mol object at 0x13580d930>
+5 <rdkit.Chem.rdchem.Mol object at 0x13580ea40>
+6 <rdkit.Chem.rdchem.Mol object at 0x13580ca50>
+7 <rdkit.Chem.rdchem.Mol object at 0x13580fbc0>
+8 <rdkit.Chem.rdchem.Mol object at 0x13580f4c0>
+9 <rdkit.Chem.rdchem.Mol object at 0x13580f060>
+10 <rdkit.Chem.rdchem.Mol object at 0x13580c200>
+11 <rdkit.Chem.rdchem.Mol object at 0x13580d770>
+12 <rdkit.Chem.rdchem.Mol object at 0x13580de70>
+13 <rdkit.Chem.rdchem.Mol object at 0x13580e2d0>
+Name: mol, dtype: object
+
+
+
At this stage, nothing really clicked at the moment, but then I thought about how Datamol was built on top of RDKit and also how a few other cheminformatics posts I’ve read before utilised the functions in RDKit, so it was time to look deeper in RDKit to search for methods with the intended purpose in mind. I then found the SmilesWriter() method from RDKit after reading a few online references.
+
I’ve found out that:
+
+
To write multiple SMILES into a .smi file, use SmilesWriter()
+
To read a set of SMILES from a .smi file, use SmilesMolSupplier()
+
+
Acknowledgement of a useful link I’ve found online which had helped me to figure out how to save multiple SMILES strings in a .smi file.
+
+
# Figured out how to save multiple SMILES as a text file
+cpd3 = SmilesWriter('cpd3.smi')
+
+# Note: saving multiple SMILES strings from RDKit mol objects (cpd3_mols)
+for s in cpd3_mols:
+ cpd3.write(s)
+cpd3.close()
+
+
+
+
Function for saving multiple SMILES strings as a .smi file
+
So based on the ideas in the previous section, I came up with the following simple function to save multiple SMILES strings as a .smi file.
+
+
def save_smiles_strings(df, file_name):
+
+# Create a RDKit mol column in the dataframe
+ df["mol"] = df.Scaffold.apply(Chem.MolFromSmiles)
+
+# Save the "mol" column with target scaffolds as an object
+ smiles_mols = df["mol"]
+
+# Use RDKit's SmilesWriter() to write the smiles strings from mol objects
+# Specify file name for .smi file, which will be stored in the working directory
+ smiles = SmilesWriter(f"{file_name}.smi")
+
+# Use a for loop to iterate through each SMILES string in the dataframe
+for s in smiles_mols:
+ smiles.write(s)
+ smiles.close()
+
+
+
+
Testing on the function
+
Here I used one of the dataframes I’ve saved earlier, cefe_scaffolds, to test this function on saving multiple SMILES into a file. Since cefe_scaffolds dataframe was a Polars dataframe from earlier, it needed to be converted into a Pandas dataframe in order to be compatible with RDKit, which was used in the function.
+
+
# Convert Polars dataframe into a Pandas one
+cefe_scaffolds = cefe_scaffolds.to_pandas()
+
+# Running function on cefe_scaffolds dataframe
+# First parameter - dataframe to be used
+# Second parameter - file name for the SMILES strings saved
+save_smiles_strings(cefe_scaffolds, "cefe")
+
+
A .smi file with the name “cefe.smi” should appear in the working directory after running the function.
+
Now, the next stage would be to parse these SMILES strings and save them as a list. I actually worked backwards here where I looked into the Pandas.query() method first, and looked into options I could have on reading and checking for matches in multiple strings. To be able to read multiple strings in one go, a list would be suitable to carry out the matching queries (note: in the scaffold_finder library, this dataframe query method was also used in its “find_scaffolds” and “get_molecules_with_scaffold” functions).
+
An example of Pandas.query() tests:
+
+
# Using the test dataset from earlier - list of top two scaffolds with highest frequency of occurrences from ChEMBL dataset
+count_top_scaffold
# To demonstrate that querying the two top scaffolds
+# will bring back all the anti-bacterials with the same scaffold
+match_df = mol_df.query("Scaffold in @count_top_scaffold")
+match_df
+
+
+
+
+
+
+
+
+
Scaffold
+
NumAtoms
+
NumRgroupgs
+
Name
+
SMILES
+
+
+
+
+
102
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
30
+
1
+
sarecycline
+
CON(C)Cc1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C...
+
+
+
128
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
+
29
+
2
+
sarecycline
+
CON(C)Cc1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C...
+
+
+
92
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
+
29
+
3
+
tigecycline
+
CN(C)c1cc(NC(=O)CNC(C)(C)C)c(O)c2c1C[C@H]1C[C@...
+
+
+
115
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
+
29
+
3
+
demeclocycline
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
+
+
129
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
30
+
2
+
demeclocycline
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
+
+
39
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
30
+
1
+
minocycline
+
CN(C)c1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C(O...
+
+
+
65
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
+
29
+
2
+
minocycline
+
CN(C)c1ccc(O)c2c1C[C@H]1C[C@H]3[C@H](N(C)C)C(O...
+
+
+
115
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
+
29
+
3
+
tetracycline
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
+
+
129
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
30
+
2
+
tetracycline
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
+
+
171
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
+
29
+
3
+
eravacycline
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
+
+
201
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
30
+
2
+
eravacycline
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
+
+
172
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
30
+
3
+
oxytetracycline
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@@]2(O)C(O)=C...
+
+
+
92
+
CN(C)[C@@H]1C(O)=C(C(N)=O)C(=O)[C@H]2C(O)=C3C(...
+
29
+
3
+
omadacycline
+
CN(C)c1cc(CNCC(C)(C)C)c(O)c2c1C[C@H]1C[C@H]3[C...
+
+
+
+
+
+
+
Here, all tetracycline antibiotics were brought up in the resultant dataframe.
+
As an aside from what I wanted to do, I also learnt a small trick about how to get the number of atoms in the file with multiple SMILES strings.
+
+
# Sample use of SmilesMolSupplier & GetNumAtoms()
+suppl = SmilesMolSupplier('cpd3.smi')
+
+nMols =len(suppl)
+
+for i inrange(nMols):
+
+ a = suppl[i].GetNumAtoms()
+print(a)
Now, back to where I was meant to continue working, I wanted to convert these SMILES strings into RDKit molecules first.
+
+
# Reading cpd3.smi SMILES strings in text file as RDKit mol objects
+suppl = SmilesMolSupplier("cpd3.smi")
+suppl
+
+
<rdkit.Chem.rdmolfiles.SmilesMolSupplier at 0x134bf5c10>
+
+
+
This was followed by converting the “mol” objects into SMILES strings, so that we could save each SMILES string into a list.
+
+
# Initialise an empty list
+list= []
+
+for mol in suppl:
+# Convert RDKit mol objects into SMILES strings
+ m = Chem.MolToSmiles(mol)
+# Add each SMILES read from filename.smi into the empty list
+list.append(m)
+
+list
Function for converting .smi file into a list to query and match scaffolds of interests
+
I then came up with the next function that would feed multiple scaffolds into a Pandas.query() to match the strings, meaning we could compare the scaffolds with each other in a dataframe.
+
+
def query_scaffolds_via_smiles(filename):
+
+# Initialise an empty list
+list= []
+# Use SmilesMolSupplier() from RDKit to read in the SMILES strings stored in .smi file
+ suppl = SmilesMolSupplier(filename)
+# Use a for loop to iterate through the SMILES strings
+for mol in suppl:
+# Convert RDKit mol objects into SMILES strings
+ m = Chem.MolToSmiles(mol)
+# Add each SMILES read from filename.smi into the empty list
+list.append(m)
+# Compare the SMILES with the scaffold column in dataframe
+ scaffold_match_df = mol_df.query("Scaffold in @list")
+
+return scaffold_match_df
+
+
+
+
Testing on the function
+
Below was a test for this query_scaffolds_via_smiles() function using the previously made “cpd3.smi” file.
+
To show that compound 3 scaffolds literally only existed in compound 3 and not in any other prescription-only anti-bacterials based on the ChEMBL-extracted anti-infective dataset only (note: other sources not checked at this stage).
+
+
# Testing query_scaffolds_via_smiles() function
+query_scaffolds_via_smiles("cpd3.smi")
+
+
+
+
+
+
+
+
+
Scaffold
+
NumAtoms
+
NumRgroupgs
+
Name
+
SMILES
+
+
+
+
+
0
+
O=CC1CCCC(Cc2ccc(O)cc2)C1=O
+
17
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
4
+
O=CC1CCCC(Cc2ccccc2)C1=O
+
16
+
2
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
5
+
CC(=O)C1CCCC(Cc2ccc(O)cc2)C1=O
+
18
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
8
+
CC(=O)C1CCCC(Cc2ccccc2)C1=O
+
17
+
2
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
9
+
CCC(=O)C1CCCC(Cc2ccc(O)cc2)C1=O
+
19
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
11
+
CCC(=O)C1CCCC(Cc2ccccc2)C1=O
+
18
+
2
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
12
+
O=C(CCc1ccccc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
25
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
13
+
O=C1CCCCC1C(=O)CCc1ccccc1
+
17
+
2
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
14
+
CC1CCCC(C(=O)CCc2ccccc2)C1=O
+
18
+
2
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
15
+
O=C(CCc1ccccc1)C1CCCC(Cc2ccccc2)C1=O
+
24
+
2
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
16
+
O=C1CCCCC1C(=O)CCc1ccc(O)cc1
+
18
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
19
+
CC1CCCC(C(=O)CCc2ccc(O)cc2)C1=O
+
19
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
21
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccccc2)C1=O
+
25
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
22
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
26
+
1
+
Compound_3
+
O=C(CCc1ccc(O)cc1)C1CCCC(Cc2ccc(O)cc2)C1=O
+
+
+
+
+
+
+
Then I also tested on “cefe.smi” created before.
+
+
# Test on cefe.smi
+query_scaffolds_via_smiles("cefe.smi")
+
+
+
+
+
+
+
+
+
Scaffold
+
NumAtoms
+
NumRgroupgs
+
Name
+
SMILES
+
+
+
+
+
0
+
C[N+]1(CC2=C(C(=O)[O-])N3C(=O)[C@@H](NC(=O)/C(...
+
31
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
1
+
C[N+]1(CC2=CN3C(=O)[C@@H](NC(=O)C(=NO)c4csc(N)...
+
28
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
2
+
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=CCS[C@H]23)cs1
+
21
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
3
+
CC1=CN2C(=O)[C@@H](NC(=O)C(=NO)c3csc(N)n3)[C@H...
+
22
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
4
+
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=C(C[NH+]4CCC...
+
27
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
5
+
C[N+]1(CC2=CN3C(=O)[C@@H](NC(=O)C=NO)[C@H]3SC2...
+
22
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
6
+
C[N+]1(CC2=CN3C(=O)[C@@H](NC(=O)C(=NO)c4cscn4)...
+
27
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
7
+
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C(C(=O)[O-])=C...
+
24
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
8
+
O=C([O-])C1=CCS[C@@H]2[C@H](NC(=O)C(=NO)c3cscn...
+
23
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
9
+
CC1=C(C(=O)[O-])N2C(=O)[C@@H](NC(=O)C(=NO)c3cs...
+
25
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
10
+
CC1=C(C(=O)[O-])N2C(=O)[C@@H](NC(=O)C=NO)[C@H]...
+
19
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
11
+
CC1=C(C(=O)[O-])N2C(=O)[C@@H](NC(=O)C(=NO)c3cs...
+
24
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
12
+
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C(C(=O)[O-])=C...
+
30
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
13
+
O=C(C=NO)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[NH+]3...
+
24
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
14
+
O=C([O-])C1=C(C[NH+]2CCCC2)CS[C@@H]2[C@H](NC(=...
+
29
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
15
+
C[N+]1(CC2=C(C(=O)[O-])N3C(=O)[C@@H](NC(=O)C=N...
+
25
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
16
+
C[N+]1(CC2=C(C(=O)[O-])N3C(=O)[C@@H](NC(=O)C(=...
+
30
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
17
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C=C(C[N+]3(C)CCCC3...
+
29
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
18
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=CCS[C@H]12)c1csc(N)n1
+
22
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
19
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=CCS[C@H]12)c1cscn1
+
21
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
20
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cs...
+
23
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
21
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cscn1
+
22
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
22
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C[NH+]3CCCC3)CS[...
+
28
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
23
+
CON=CC(=O)N[C@@H]1C(=O)N2C=C(C[NH+]3CCCC3)CS[C...
+
22
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
24
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C[NH+]3CCCC3)CS[...
+
27
+
3
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
25
+
CON=CC(=O)N[C@@H]1C(=O)N2C=C(C[N+]3(C)CCCC3)CS...
+
23
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
26
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C[N+]3(C)CCCC3)C...
+
28
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
27
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=CCS[C...
+
25
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
28
+
CON=C(C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=CCS[C@H...
+
25
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
30
+
CON=C(C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=CCS[C@H...
+
24
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
31
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C)C...
+
26
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
32
+
CON=C(C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C)CS[...
+
26
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
33
+
CON=CC(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C)CS[C...
+
20
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
34
+
CON=C(C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C)CS[...
+
25
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
35
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
31
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
36
+
CON=CC(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[NH+]...
+
25
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
37
+
CON=C(C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[NH+...
+
30
+
2
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
38
+
CO/N=C\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N+...
+
26
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
39
+
CON=CC(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N+]3...
+
26
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
40
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
31
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
41
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
32
+
1
+
cefepime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=C(C[N...
+
+
+
2
+
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=CCS[C@H]23)cs1
+
21
+
3
+
cefixime
+
C=CC1=C(C(=O)O)N2C(=O)[C@@H](NC(=O)/C(=N\OCC(=...
+
+
+
3
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=CCS[C@H]12)c1csc(N)n1
+
22
+
3
+
cefixime
+
C=CC1=C(C(=O)O)N2C(=O)[C@@H](NC(=O)/C(=N\OCC(=...
+
+
+
2
+
CC1=CN2C(=O)[C@@H](NC(=O)C(=NO)c3csc(N)n3)[C@H...
+
22
+
3
+
ceftriaxone
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(CSc3nc...
+
+
+
24
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cs...
+
23
+
2
+
ceftriaxone
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(CSc3nc...
+
+
+
25
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cscn1
+
22
+
3
+
ceftriaxone
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(CSc3nc...
+
+
+
1
+
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=CCS[C@H]23)cs1
+
21
+
2
+
cefdinir
+
C=CC1=C(C(=O)O)N2C(=O)[C@@H](NC(=O)/C(=N\O)c3c...
+
+
+
2
+
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C=CCS[C@H]23)cs1
+
21
+
3
+
cefotaxime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
+
+
+
3
+
CC1=CN2C(=O)[C@@H](NC(=O)C(=NO)c3csc(N)n3)[C@H...
+
22
+
3
+
cefotaxime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
+
+
+
19
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=CCS[C@H]12)c1csc(N)n1
+
22
+
2
+
cefotaxime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
+
+
+
20
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=CCS[C@H]12)c1cscn1
+
21
+
3
+
cefotaxime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
+
+
+
21
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cs...
+
23
+
2
+
cefotaxime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
+
+
+
22
+
CON=C(C(=O)N[C@@H]1C(=O)N2C=C(C)CS[C@H]12)c1cscn1
+
22
+
3
+
cefotaxime
+
CO/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)O)=C(COC(C)...
+
+
+
33
+
CC1=CN2C(=O)[C@@H](NC(=O)C(=NO)c3csc(N)n3)[C@H...
+
22
+
3
+
ceftazidime
+
CC(C)(O/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=...
+
+
+
36
+
Nc1nc(C(=NO)C(=O)N[C@@H]2C(=O)N3C(C(=O)[O-])=C...
+
24
+
2
+
ceftazidime
+
CC(C)(O/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=...
+
+
+
37
+
O=C([O-])C1=CCS[C@@H]2[C@H](NC(=O)C(=NO)c3cscn...
+
23
+
3
+
ceftazidime
+
CC(C)(O/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=...
+
+
+
38
+
CC1=C(C(=O)[O-])N2C(=O)[C@@H](NC(=O)C(=NO)c3cs...
+
25
+
2
+
ceftazidime
+
CC(C)(O/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=...
+
+
+
39
+
CC1=C(C(=O)[O-])N2C(=O)[C@@H](NC(=O)C(=NO)c3cs...
+
24
+
3
+
ceftazidime
+
CC(C)(O/N=C(\C(=O)N[C@@H]1C(=O)N2C(C(=O)[O-])=...
+
+
+
+
+
+
+
Only cephalosporins were brought up as the result from this scaffold query, which matched with the antibiotic class of cefepime.
+
+
+
+
+
+
Some answers
+
There were no other prescription-only anti-bacterials from ChEMBL database with the same scaffold as compound 3 after comparing the SMILES strings between these selected scaffolds only. This was only limited to the dataset obtained from ChEMBL at this stage, with compound indications limited to anti-infectives for now. This might imply that the scaffold of compound 3 could be considered novel when comparing with molecules with similar indications. Obviously, this was too preliminary to confirm anything substantial, since there were no in vivo tests on finding out the efficacy, safety and toxicity of compound 3 apart from the in vitro experimental results mentioned in the paper. It probably would provide some ideas for scaffold hopping in hit compounds, or functional group (R-group) comparisons when looking for new compounds for synthesis. Overall, it was interesting to have a re-visit on this work using a more cheminformatics approach.
+
+
+
+
Afterthoughts
+
What I wanted to achieve in this post were:
+
+
To familiarise myself with Datamol and scaffold_finder libraries
+
To use Polars dataframe library for initial data wrangling along with Datamol Python library, and the later trial of scaffold_finder library. Using Polars would be a small degree only, as Datamol was likely written with Pandas in mind mostly (based on RDKit), while Pandas was also the more commonly used dataframe libray in many cheminformatics packages. Some people might prefer to stick with Pandas all the way, which I agree, but I’d just wanted to use Polars for the initial data wrangling only as I’ve been using it more lately
+
To reveal my thought process on building simple cheminformatics-related functions (this was unplanned, but kind of evolved while working on this post)
+
To show some evidence of my own growth from computational and medicinal chemistry with no code, to using data science tools with Python code to help guiding drug discovery projects
+
To mention that experimental validations will always be crucial for computational predictions, and since I had some experimental results from the paper, I thought to accompany it with some computational findings here
+
+
I hope I’ve at least achieved some these points in this post if not all.
+
Thanks for reading and looking forward to comments if any.
+
+
+
+
+
+
Footnotes
+
+
+
Simplified Molecular Input Line Entry Systems↩︎
+
SELF-referencIng Embedded Strings↩︎
+
International Chemical Identifier↩︎
+
+
]]>
+ RDKit
+ Datamol
+ Python
+ Pandas
+ Polars
+ Cheminformatics
+ https://jhylin.github.io/Data_in_life_blog/posts/14_Scaffolds_in_small_molecules/chembl_anti-inf_data_prep_current.html
+ Wed, 05 Jul 2023 12:00:00 GMT
+
+
+ Shinylive app in Python
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_app_embed_pyodide_http.html
+
+
Quick update
+
I’ve changed the way of importing a local text/csv file from manually copying-and-pasting to using pyodide.http.open_url() in the shinylive app, which works great and avoids the clumsy manual file input. I couldn’t quite grasp the code back then, but managed to get it this time when I re-visited the problem, while also figured out that I could use the raw content link for the file from my GitHub repository. This method was also inspired by the same user answering the query in this GitHub discussion. So I’ve basically trialled both ways as suggested, which have all worked.
+
Note: if importing binary files, use pyodide.http.pyfetch() instead - check out Pyodide for details and latest changes.
+
+
+
Shinylive app in action
+
Note: it may take a few minutes to load the app (code provided at the top, with app at the bottom).
+
+
#| standalone: true
+#| components: [editor, viewer]
+#| layout: vertical
+#| viewerHeight: 420
+
+## file: app.py
+# ***Import all libraries or packages needed***
+# Import shiny ui, app
+from shiny import ui, App
+# Import shinywidgets
+from shinywidgets import output_widget, render_widget
+# Import shinyswatch to add themes
+#import shinyswatch
+# Import plotly express
+import plotly.express as px
+# Import pandas
+import pandas as pd
+# Import pyodide http - for importing file via URL
+import pyodide.http
+from pyodide.http import open_url
+
+
+# ***Specify data source***
+# Using pyodide.http.open_url
+df = pd.read_csv(open_url('https://raw.githubusercontent.com/jhylin/Data_in_life_blog/main/posts/13_Shiny_app_python/pc_cov_pd.csv'))
+
+
+# User interface---
+# Add inputs & outputs
+app_ui = ui.page_fluid(
+ # Add theme - seems to only work in VS code and shinyapps.io
+ #shinyswatch.theme.superhero(),
+ # Add heading
+ ui.h3("Molecular properties of compounds used in COVID-19 clinical trials"),
+ # Place selection boxes & texts in same row
+ ui.row(
+ # Divide the row into two columns
+ # Column 1 - selection drop-down boxes x 2
+ ui.column(
+ 4, ui.input_select(
+ # Specify x variable input
+ "x", label = "x axis:",
+ choices = ["Partition coefficients",
+ "Complexity",
+ "Heavy atom count",
+ "Hydrogen bond donor count",
+ "Hydrogen bond acceptor count",
+ "Rotatable bond count",
+ "Molecular weight",
+ "Exact mass",
+ "Polar surface area",
+ "Total atom stereocenter count",
+ "Total bond stereocenter count"],
+ ),
+ ui.input_select(
+ # Specify y variable input
+ "y", label = "y axis:",
+ choices = ["Partition coefficients",
+ "Complexity",
+ "Heavy atom count",
+ "Hydrogen bond donor count",
+ "Hydrogen bond acceptor count",
+ "Rotatable bond count",
+ "Molecular weight",
+ "Exact mass",
+ "Polar surface area",
+ "Total atom stereocenter count",
+ "Total bond stereocenter count"]
+ )),
+ # Column 2 - add texts regarding plots
+ ui.column(
+ 8,
+ ui.p("Select different molecular properties as x and y axes to produce a scatter plot."),
+ ui.tags.ul(
+ ui.tags.li(
+ """
+ Part_coef_group means groups of partition coefficient (xlogp) as shown in the legend on the right"""
+ ),
+ ui.tags.li(
+ """
+ Toggle each partition coefficient category by clicking on the group names"""
+ ),
+ ui.tags.li(
+ """
+ Hover over each data point to see compound name and relevant molecular properties"""
+ )
+ )),
+ # Output as a widget (interactive plot)
+ output_widget("my_widget"),
+ # Add texts for data source
+ ui.row(
+ ui.p(
+ """
+ Data curated by PubChem, accessed from: https://pubchem.ncbi.nlm.nih.gov/#tab=compound&query=covid-19%20clinicaltrials (last access date: 30th Apr 2023)"""
+ )
+ )
+ )
+)
+
+
+# Server---
+# Add plotting code within my_widget function within the server function
+def server(input, output, session):
+ @output
+ @render_widget
+ def my_widget():
+ fig = px.scatter(
+ df, x = input.x(), y = input.y(),
+ color = "Part_coef_group",
+ hover_name = "Compound name"
+ )
+ fig.layout.height = 400
+ return fig
+
+# Combine UI & server into Shiny app
+app = App(app_ui, server)
+
+
+
+
+
+
+ ]]>
+ Python
+ Shiny
+ Pandas
+ Plotly
+ PubChem
+ Cheminformatics
+ https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_app_embed_pyodide_http.html
+ Tue, 23 May 2023 12:00:00 GMT
+
+
+ Shinylive app in Python
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_app_embed.html
+
+
Coding for Shinylive app in Python
+
The entire code for this Shiny app in Python were written in VS Code initially. When I was looking for places to deploy the app, I then migrated the code to RStudio IDE (note: recently I saw Python code in Quarto documents being used in VS Code after I’ve deployed this app, so this might be another option).
+
For data preparation stage, please visit this post for details.
+
+
+
Trialling open-source large-language models
+
There have been a lot of hypes surrounding large-language models (LLMs) or generative pre-trained transformers (GPTs). I’m still somewhat both reserved and excited about them at present. The area I’m most concerned with is where are the relevant laws, regulations and ethics on using these tools in public domains, whether for private or commercial uses, and will they be country-specific or globally streamlined? While I have these questions in mind, I’m not denying the fact that they can be useful for a few well-known purposes in coding, such as requesting for regex code templates for text cleaning or asking for code snippets while programmers are busy with several tasks at the same time.
+
I thought to trial out the open-source LLMs a little bit when I was working on this app, since they were the open and transparent versions with potentials for personal or commercial uses. So I’ve tested about 3 different open-source LLMs, which were H2OGPT, HuggingChat and StableLM. I decided to only give minimal amount of prompts (I’ve given H2OGPT three prompts, and only one prompt each for either HuggingChat and StableLM), and the question was framed around providing a code outline for building a Shiny app using Python and Polars dataframe libary, and see what answers they could each provide.
+
All of them produced answers that were close to what I’ve asked for, i.e. I could see a code outline for a Shiny app.py file at first but it was more like R code, with one case mixing both Python and R in the code concurrently. However, none of them actually reached exactly how I’ve asked them to provide, perhaps if I’ve given more prompts then they might manage to provide answers closer to the request. Also, none of them were able to pick up the more recent Polars dataframe library (which were probably more prominent from 2021 onwards) and also Shiny in Python (which was also quite new as well, only out of alpha phase recently). All of them only showed code using Pandas dataframe libray, even though I did mention Polars in all of the question prompts. Once I get to use more of these open-source LLMs in the future, which currently are gaining more traction from what I’ve read online, I think I might try to write another post about them later.
+
+
+
+
Using Shiny in Python documentations
+
After trialling open-source LLMs a bit to help with the coding part (although not as helpful as I first imagined, but they still somewhat provided a rough code framework for Shiny apps), I completed the app.py script in the end by using mainly documents from Shiny for Python website. I still think thorough documentations for any products are still fundamental and inevitable, because these documentations will be sources to provide high-quality data for training these LLMs which means they’ll be of higher quality too.
+
Several links I’ve used and found to be very informative:
+
+
“Shiny for Python out of alpha” - quick summary on recent status update and new features such as shinyswatch and shinywidgets on Shiny for Python
+
“Quickstart for R users” - a very useful link for people who are R users or already familiar with Shiny apps in R, and would like to build Shiny apps in Python
After building the app in a workable condition, I started looking at where and how to deploy this Shiny app written in Python. Initially I used the easiest method as stated from the Shinylive link above, which was to deploy on Shinylive editor. One drawback for this was that the application URL was extremely long as the URL hash would store the entire app code inside. However, this was indeed one of the simplest ways to share the apps with others quickly, by simply providing the URL link with the intended audience.
+
I then went on to try another method where I spent one afternoon trying to figure out that I had to add an install_certificate.command for my operating system since I installed Python by using Homebrew in the first place (which some clever users might already know to avoid…), so that I could resolve the SSL certificate issue if I’m trying to connect with Shinyapps.io.
+
I also tried to follow another method from an earlier time which suggested to deploy the app directly on GitHub Pages, but since then the code was changed and updated to a different version and the old method no longer worked as nicely as mentioned (a lot of the code for Shiny in Python can still be experimental, and can change drastically, so check the sources to see the latest updates). I then stumbled upon another GitHub repository from the Quarto team at Posit/RStudio and looked into the possibility and tested it. Eventually, I settled on this method - embedding the app in a Quarto document, which provided both code and app itself on this very same webpage down below.
+
One of the downsides of the following code that I couldn’t quite get rid of yet was the poor file importation style. for loading a .csv file locally. I’ve added code annotations as comments to explain what I’ve done. The solution to import .csv file into app.py for the embedding version was from this discussion in the repository (thanks to that particular user who found this hacky way to do it). However, the plus side was that coding for the rest of the app was not bad at all and worked seamlessly. I’ll try to follow up later on whether importing .csv file for app embedding could be easier, perhaps this might be something the Quarto team is working right now.
+
The second downside was that shinyswatch package might not be functional yet for this embedding method (but most likely would work if deploying apps to shinyapps.io). This meant there would be no background visual themes yet for apps embedded in Quarto docs, but hopefully this would be possible in the future. Overall, I was amazed at how easy it was to build, deploy and embed a Shiny app in Python in a Quarto document.
+
Note: if using Shinylive editor, make sure to include a requirement.txt file if using extra Python packages such as Pandas or Plotly. For embedding apps in Quarto docs, this appears not to be compulsory and only optional when I tried it down below.
+
Code for this post (.qmd document) is available here.
+
+
+
Shinylive app in action
+
Note: it may take a few minutes to load the app (code provided at the top, with app at the bottom).
+
Update on 24/5/23: to avoid manual file input, see this updated post which uses pyodide.http.open_url() method (code change in editor section).
+
+
#| standalone: true
+#| components: [editor, viewer]
+#| layout: vertical
+#| viewerHeight: 420
+
+## file: app.py
+# ***Import all libraries or packages needed***
+# Import shiny ui, app
+from shiny import ui, App
+# Import shinywidgets
+from shinywidgets import output_widget, render_widget
+# Import shinyswatch to add themes
+#import shinyswatch
+# Import plotly express
+import plotly.express as px
+# Import pandas
+import pandas as pd
+from pathlib import Path
+
+
+# User interface---
+# Add inputs & outputs
+app_ui = ui.page_fluid(
+ # Add theme - seems to only work in VS code and shinyapps.io
+ #shinyswatch.theme.superhero(),
+ # Add heading
+ ui.h3("Molecular properties of compounds used in COVID-19 clinical trials"),
+ # Place selection boxes & texts in same row
+ ui.row(
+ # Divide the row into two columns
+ # Column 1 - selection drop-down boxes x 2
+ ui.column(
+ 4, ui.input_select(
+ # Specify x variable input
+ "x", label = "x axis:",
+ choices = ["Partition coefficients",
+ "Complexity",
+ "Heavy atom count",
+ "Hydrogen bond donor count",
+ "Hydrogen bond acceptor count",
+ "Rotatable bond count",
+ "Molecular weight",
+ "Exact mass",
+ "Polar surface area",
+ "Total atom stereocenter count",
+ "Total bond stereocenter count"],
+ ),
+ ui.input_select(
+ # Specify y variable input
+ "y", label = "y axis:",
+ choices = ["Partition coefficients",
+ "Complexity",
+ "Heavy atom count",
+ "Hydrogen bond donor count",
+ "Hydrogen bond acceptor count",
+ "Rotatable bond count",
+ "Molecular weight",
+ "Exact mass",
+ "Polar surface area",
+ "Total atom stereocenter count",
+ "Total bond stereocenter count"]
+ )),
+ # Column 2 - add texts regarding plots
+ ui.column(
+ 8,
+ ui.p("Select different molecular properties as x and y axes to produce a scatter plot."),
+ ui.tags.ul(
+ ui.tags.li(
+ """
+ Part_coef_group means groups of partition coefficient (xlogp) as shown in the legend on the right"""
+ ),
+ ui.tags.li(
+ """
+ Toggle each partition coefficient category by clicking on the group names"""
+ ),
+ ui.tags.li(
+ """
+ Hover over each data point to see compound name and relevant molecular properties"""
+ )
+ )),
+ # Output as a widget (interactive plot)
+ output_widget("my_widget"),
+ # Add texts for data source
+ ui.row(
+ ui.p(
+ """
+ Data curated by PubChem, accessed from: https://pubchem.ncbi.nlm.nih.gov/#tab=compound&query=covid-19%20clinicaltrials (last access date: 30th Apr 2023)"""
+ )
+ )
+ )
+)
+
+
+# Server---
+# Add plotting code within my_widget function within the server function
+def server(input, output, session):
+ @output
+ @render_widget
+ def my_widget():
+ fig = px.scatter(
+ df, x = input.x(), y = input.y(),
+ color = "Part_coef_group",
+ hover_name = "Compound name"
+ )
+ fig.layout.height = 400
+ return fig
+
+# Combine UI & server into Shiny app
+app = App(app_ui, server)
+
+
+# ***Specify data source***
+# --Not the best approach yet but works for now--
+# Currently this work-around only suits small to medium-size dataset
+# Not ideal for large dataset for sure
+# To load file locally use the following code
+infile = Path(__file__).parent / "pc_cov_pd.csv"
+df = pd.read_csv(infile)
+
+# Then manually paste in csv/txt file below
+## file: pc_cov_pd.csv
+,Compound name,Molecular weight,Polar surface area,Complexity,Partition coefficients,Heavy atom count,Hydrogen bond donor count,Hydrogen bond acceptor count,Rotatable bond count,Exact mass,Monoisotopic mass,Formal charge,Covalently-bonded unit count,Isotope atom count,Total atom stereocenter count,Defined atom stereocenter count,Undefined atoms stereocenter count,Total bond stereocenter count,Defined bond stereocenter count,Undefined bond stereocenter count,Part_coef_group
+0,Calcitriol,416.6,60.7,688.0,5.1,30,3,3,6,416.329,416.329,0,1,0,6,6,0,2,2,0,Larger than 6
+1,Ubiquinol,865.4,58.9,1600.0,20.2,63,2,4,31,864.7,864.7,0,1,0,0,0,0,9,9,0,Larger than 6
+2,Glutamine,146.14,106.0,146.0,-3.1,10,3,4,4,146.069,146.069,0,1,0,1,1,0,0,0,0,Between -11 and 5
+3,Aspirin,180.16,63.6,212.0,1.2,13,1,4,3,180.042,180.042,0,1,0,0,0,0,0,0,0,Between -11 and 5
+4,1-Methylnicotinamide,137.16,47.0,136.0,-0.1,10,1,1,1,137.071,137.071,1,1,0,0,0,0,0,0,0,Between -11 and 5
+5,Losartan,422.9,92.5,520.0,4.3,30,2,5,8,422.162,422.162,0,1,0,0,0,0,0,0,0,Between -11 and 5
+6,Vitamin E,430.7,29.5,503.0,10.7,31,1,2,12,430.381,430.381,0,1,0,3,3,0,0,0,0,Larger than 6
+7,Nicotinamide,122.12,56.0,114.0,-0.4,9,1,2,1,122.048,122.048,0,1,0,0,0,0,0,0,0,Between -11 and 5
+8,Adenosine,267.24,140.0,335.0,-1.1,19,4,8,2,267.097,267.097,0,1,0,4,4,0,0,0,0,Between -11 and 5
+9,Inosine,268.23,129.0,405.0,-1.3,19,4,7,2,268.081,268.081,0,1,0,4,4,0,0,0,0,Between -11 and 5
+10,Nicotinamide riboside,255.25,117.0,314.0,-1.8,18,4,5,3,255.098,255.098,1,1,0,4,4,0,0,0,0,Between -11 and 5
+11,Dimethyl Fumarate,144.12,52.6,141.0,0.7,10,0,4,4,144.042,144.042,0,1,0,0,0,0,1,1,0,Between -11 and 5
+12,Inosinic acid,348.21,176.0,555.0,-3.0,23,5,10,4,348.047,348.047,0,1,0,4,4,0,0,0,0,Between -11 and 5
+13,Acetyl-L-carnitine,203.24,66.4,214.0,0.4,14,0,4,5,203.116,203.116,0,1,0,1,1,0,0,0,0,Between -11 and 5
+14,Camostat,398.4,137.0,602.0,1.1,29,2,6,9,398.159,398.159,0,1,0,0,0,0,0,0,0,Between -11 and 5
+15,Estradiol,272.4,40.5,382.0,4.0,20,2,2,0,272.178,272.178,0,1,0,5,5,0,0,0,0,Between -11 and 5
+16,Aspartic Acid,133.1,101.0,133.0,-2.8,9,3,5,3,133.038,133.038,0,1,0,1,1,0,0,0,0,Between -11 and 5
+17,Ribavirin,244.2,144.0,304.0,-1.8,17,4,7,3,244.081,244.081,0,1,0,4,4,0,0,0,0,Between -11 and 5
+18,Angiotensin (1-7),899.0,380.0,1660.0,-3.0,64,12,14,25,898.466,898.466,0,1,0,8,8,0,0,0,0,Between -11 and 5
+19,alpha-Maltose,342.3,190.0,382.0,-4.7,23,8,11,4,342.116,342.116,0,1,0,10,10,0,0,0,0,Between -11 and 5
+20,Ambrisentan,378.4,81.5,475.0,3.8,28,1,6,7,378.158,378.158,0,1,0,1,1,0,0,0,0,Between -11 and 5
+21,Ciclosporin,1202.6,279.0,2330.0,7.5,85,5,12,15,1201.84,1201.84,0,1,0,12,12,0,1,1,0,Larger than 6
+22,Ergocalciferol,396.6,20.2,678.0,7.4,29,1,1,5,396.339,396.339,0,1,0,6,6,0,3,3,0,Larger than 6
+23,Docosahexaenoic Acid,328.5,37.3,462.0,6.2,24,1,2,14,328.24,328.24,0,1,0,0,0,0,6,6,0,Larger than 6
+24,Tacrolimus,804.0,178.0,1480.0,2.7,57,3,12,7,803.482,803.482,0,1,0,14,14,0,2,2,0,Between -11 and 5
+25,Budesonide,430.5,93.1,862.0,2.5,31,2,6,4,430.236,430.236,0,1,0,9,8,1,0,0,0,Between -11 and 5
+26,Calcifediol,400.6,40.5,655.0,6.2,29,2,2,6,400.334,400.334,0,1,0,5,5,0,2,2,0,Larger than 6
+27,Cepharanthine,606.7,61.9,994.0,6.5,45,0,8,2,606.273,606.273,0,1,0,2,2,0,0,0,0,Larger than 6
+28,Cholecalciferol,384.6,20.2,610.0,7.9,28,1,1,6,384.339,384.339,0,1,0,5,5,0,2,2,0,Larger than 6
+29,Coenzyme Q10,863.3,52.6,1840.0,19.4,63,0,4,31,862.684,862.684,0,1,0,0,0,0,9,9,0,Larger than 6
+30,Tacrolimus monohydrate,822.0,179.0,1480.0,,58,4,13,7,821.493,821.493,0,2,0,14,14,0,2,2,0,Larger than 6
+31,Alisporivir,1216.6,279.0,2360.0,7.9,86,5,12,15,1215.86,1215.86,0,1,0,13,13,0,1,1,0,Larger than 6
+32,Pacritinib,472.6,68.7,644.0,3.8,35,1,7,4,472.247,472.247,0,1,0,0,0,0,1,1,0,Between -11 and 5
+33,Topotecan,421.4,103.0,867.0,0.5,31,2,7,3,421.164,421.164,0,1,0,1,1,0,0,0,0,Between -11 and 5
+34,Voclosporin,1214.6,279.0,2380.0,7.9,86,5,12,16,1213.84,1213.84,0,1,0,12,12,0,1,1,0,Larger than 6
+35,"(1R,4S,5'S,6R,6'R,8R,10E,12S,13S,14E,16E,20R,21R,24S)-6'-[(2R)-Butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,3,0,Between -11 and 5
+36,Oxytocin,1007.2,450.0,1870.0,-2.6,69,12,15,17,1006.44,1006.44,0,1,0,9,9,0,0,0,0,Between -11 and 5
+37,"(1R,4S,6R,10E,14E,16E,21R)-6'-butan-2-yl-21,24-dihydroxy-12-[(2R,4S,6S)-5-[(2S,4S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,10,10,3,3,0,Between -11 and 5
+38,"Manganese, dichloro((4aS,13aS,17aS,21aS)-1,2,3,4,4a,5,6,12,13,13a,14,15,16,17,17a,18,19,20,21,21a-eicosahydro-7,11-nitrilo-7H-dibenzo(b,H)-5,13,18,21-tetraazacycloheptadecine-kappaN5,kappaN13,kappaN18,kappaN21,kappaN22)-, (pb-7-11-2344'3')-",483.4,61.0,381.0,,29,4,7,0,482.165,482.165,0,4,0,4,4,0,0,0,0,Larger than 6
+39,Ivermectin B1a,875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,3,0,Between -11 and 5
+40,Dipyridamole,504.6,145.0,561.0,0.7,36,4,12,12,504.317,504.317,0,1,0,0,0,0,0,0,0,Between -11 and 5
+41,Tetrandrine,622.7,61.9,979.0,6.4,46,0,8,4,622.304,622.304,0,1,0,2,2,0,0,0,0,Larger than 6
+42,Sirolimus,914.2,195.0,1760.0,6.0,65,3,13,6,913.555,913.555,0,1,0,15,15,0,4,4,0,Larger than 6
+43,Iloprost,360.5,77.8,606.0,2.8,26,3,4,8,360.23,360.23,0,1,0,6,5,1,2,2,0,Between -11 and 5
+44,Ramipril,416.5,95.9,619.0,1.4,30,2,6,10,416.231,416.231,0,1,0,5,5,0,0,0,0,Between -11 and 5
+45,Prasugrel hydrochloride,409.9,74.8,555.0,,27,1,6,6,409.091,409.091,0,2,0,1,0,1,0,0,0,Larger than 6
+46,Uproleselan,1304.5,383.0,1870.0,-2.0,90,9,27,52,1303.72,1303.72,0,1,0,15,15,0,0,0,0,Between -11 and 5
+47,MET-enkephalin,573.7,225.0,847.0,-2.1,40,7,9,16,573.226,573.226,0,1,0,3,3,0,0,0,0,Between -11 and 5
+48,"(18Z)-1,14-dihydroxy-12-[(E)-1-(4-hydroxy-3-methoxycyclohexyl)prop-1-en-2-yl]-23,25-dimethoxy-13,19,21,27-tetramethyl-17-prop-2-enyl-11,28-dioxa-4-azatricyclo[22.3.1.04,9]octacos-18-ene-2,3,10,16-tetrone",804.0,178.0,1480.0,2.7,57,3,12,7,803.482,803.482,0,1,0,14,0,14,2,2,0,Between -11 and 5
+49,"(1R,4S,5'S,6R,6'R,8R,10E,13S,14E,16E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,19,1,3,3,0,Between -11 and 5
+50,Plitidepsin,1110.3,285.0,2200.0,5.7,79,4,15,15,1109.63,1109.63,0,1,0,12,12,0,0,0,0,Larger than 6
+51,"(10Z,14Z,16Z)-6'-butan-2-yl-21,24-dihydroxy-12-[5-(5-hydroxy-4-methoxy-6-methyloxan-2-yl)oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,0,20,3,3,0,Between -11 and 5
+52,"(1R,4S,5'S,6R,6'R,8R,12S,13S,20R,21R,24S)-6'-[(2R)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,0,3,Between -11 and 5
+53,Rubramin,1355.4,476.0,3220.0,,93,9,21,16,1354.57,1354.57,-3,3,0,14,14,0,3,3,0,Larger than 6
+54,"(1R,4S,5'S,6R,6'S,8R,10E,12S,13S,14E,16E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,3,0,Between -11 and 5
+55,Zidovudine,267.24,93.2,484.0,0.0,19,2,6,3,267.097,267.097,0,1,0,3,3,0,0,0,0,Between -11 and 5
+56,Resveratrol,228.24,60.7,246.0,3.1,17,3,3,2,228.079,228.079,0,1,0,0,0,0,1,1,0,Between -11 and 5
+57,Curcumin,368.4,93.1,507.0,3.2,27,2,6,8,368.126,368.126,0,1,0,0,0,0,2,2,0,Between -11 and 5
+58,"6'-Butan-2-yl-21,24-dihydroxy-12-[5-(5-hydroxy-4-methoxy-6-methyloxan-2-yl)oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,0,20,3,0,3,Between -11 and 5
+59,Regorafenib,482.8,92.4,686.0,4.2,33,3,8,5,482.077,482.077,0,1,0,0,0,0,0,0,0,Between -11 and 5
+60,Pharmakon1600-01300027,875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,17,3,3,3,0,Between -11 and 5
+61,"(1R,4S,5'S,6R,6'R,8R,10Z,12S,13S,14Z,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,2,1,Between -11 and 5
+62,"(1R,4S,5'S,6R,6'R,8R,10E,12S,13S,14E,16E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4R,5S,6S)-5-[(2S,4R,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,3,0,Between -11 and 5
+63,Methylcobalamin,1345.4,449.0,3160.0,,92,10,20,26,1344.6,1344.6,0,3,0,14,13,1,3,3,0,Larger than 6
+64,Spironolactone,416.6,85.7,818.0,2.9,29,0,5,2,416.202,416.202,0,1,0,7,7,0,0,0,0,Between -11 and 5
+65,Prednisone,358.4,91.7,764.0,1.5,26,2,5,2,358.178,358.178,0,1,0,6,6,0,0,0,0,Between -11 and 5
+66,Quercetin,302.23,127.0,488.0,1.5,22,5,7,1,302.043,302.043,0,1,0,0,0,0,0,0,0,Between -11 and 5
+67,Calderol,418.7,41.5,655.0,,30,3,3,6,418.345,418.345,0,2,0,5,5,0,2,2,0,Larger than 6
+68,Dactolisib,469.5,73.1,872.0,5.2,36,0,4,3,469.19,469.19,0,1,0,0,0,0,0,0,0,Larger than 6
+69,Enzalutamide,464.4,109.0,839.0,3.6,32,1,8,3,464.093,464.093,0,1,0,0,0,0,0,0,0,Between -11 and 5
+70,Selinexor,443.3,97.6,621.0,3.0,31,2,12,5,443.093,443.093,0,1,0,0,0,0,1,1,0,Between -11 and 5
+71,Chlorpromazine,318.9,31.8,339.0,5.2,21,0,3,4,318.096,318.096,0,1,0,0,0,0,0,0,0,Larger than 6
+72,Itraconazolum [Latin],705.6,101.0,1120.0,5.7,49,0,9,11,704.239,704.239,0,1,0,3,0,3,0,0,0,Larger than 6
+73,Loratadine,382.9,42.4,569.0,5.2,27,0,3,2,382.145,382.145,0,1,0,0,0,0,0,0,0,Larger than 6
+74,Nifedipine,346.3,110.0,608.0,2.2,25,1,7,5,346.116,346.116,0,1,0,0,0,0,0,0,0,Between -11 and 5
+75,Prazosin,383.4,107.0,544.0,2.0,28,1,8,4,383.159,383.159,0,1,0,0,0,0,0,0,0,Between -11 and 5
+76,Alizarin,240.21,74.6,378.0,3.2,18,2,4,0,240.042,240.042,0,1,0,0,0,0,0,0,0,Between -11 and 5
+77,Methylprednisolone,374.5,94.8,754.0,1.9,27,3,5,2,374.209,374.209,0,1,0,8,8,0,0,0,0,Between -11 and 5
+78,Chloroquine sulfate,418.0,111.0,390.0,,27,3,7,8,417.149,417.149,0,2,0,1,0,1,0,0,0,Larger than 6
+79,Bardoxolone methyl,505.7,84.2,1210.0,6.7,37,0,5,2,505.319,505.319,0,1,0,7,7,0,0,0,0,Larger than 6
+80,"(1R,4S,5'S,6R,6'R,8R,12S,13S,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,0,3,Between -11 and 5
+81,Heartgard-30,1736.2,340.0,3340.0,,123,6,28,15,1736.0,1735.0,0,2,0,39,34,5,6,6,0,Larger than 6
+82,Ivermectine 100 microg/mL in Acetonitrile,1736.2,340.0,3340.0,,123,6,28,15,1736.0,1735.0,0,2,0,39,39,0,6,6,0,Larger than 6
+83,Duvelisib,416.9,86.8,668.0,4.1,30,2,5,4,416.115,416.115,0,1,0,1,1,0,0,0,0,Between -11 and 5
+84,"(4S,5'S,6R,6'R,8R,10E,12S,13S,14E,16E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,19,1,3,3,0,Between -11 and 5
+85,Zotatifin,487.5,108.0,819.0,2.4,36,2,8,6,487.211,487.211,0,1,0,5,5,0,0,0,0,Between -11 and 5
+86,MaxEPA,645.0,74.6,874.0,,47,2,4,28,644.48,644.48,0,2,0,0,0,0,11,11,0,Larger than 6
+87,"(1R,4S,5'S,6R,6'R,8R,12S,13S,14E,16E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,2,1,Between -11 and 5
+88,"(1S,4S,5'S,6R,6'R,8R,10E,12S,13S,14E,20R,21R,24S)-6'-[(2S)-butan-2-yl]-21,24-dihydroxy-12-[(2R,4S,5S,6S)-5-[(2S,4S,5S,6S)-5-hydroxy-4-methoxy-6-methyloxan-2-yl]oxy-4-methoxy-6-methyloxan-2-yl]oxy-5',11,13,22-tetramethylspiro[3,7,19-trioxatetracyclo[15.6.1.14,8.020,24]pentacosa-10,14,16,22-tetraene-6,2'-oxane]-2-one",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,2,1,Between -11 and 5
+89,Indomethacin,357.8,68.5,506.0,4.3,25,1,4,4,357.077,357.077,0,1,0,0,0,0,0,0,0,Between -11 and 5
+90,Methylene Blue,319.9,43.9,483.0,,21,0,4,1,319.091,319.091,0,2,0,0,0,0,0,0,0,Larger than 6
+91,Bromocresol green,698.0,92.2,742.0,6.5,31,2,5,2,697.725,693.73,0,1,0,0,0,0,0,0,0,Larger than 6
+92,Hydroxychloroquine sulfate,434.0,131.0,413.0,,28,4,8,9,433.144,433.144,0,2,0,1,0,1,0,0,0,Larger than 6
+93,Ritonavir,720.9,202.0,1040.0,6.0,50,4,9,18,720.313,720.313,0,1,0,4,4,0,0,0,0,Larger than 6
+94,Fisetin,286.24,107.0,459.0,2.0,21,4,6,1,286.048,286.048,0,1,0,0,0,0,0,0,0,Between -11 and 5
+95,Palbociclib,447.5,103.0,775.0,1.8,33,2,8,5,447.238,447.238,0,1,0,0,0,0,0,0,0,Between -11 and 5
+96,Ruxolitinib,306.4,83.2,453.0,2.1,23,1,4,4,306.159,306.159,0,1,0,1,1,0,0,0,0,Between -11 and 5
+97,Sildenafil Citrate,666.7,250.0,1070.0,,46,5,15,12,666.232,666.232,0,2,0,0,0,0,0,0,0,Larger than 6
+98,Nintedanib,539.6,102.0,892.0,4.3,40,2,7,8,539.253,539.253,0,1,0,0,0,0,0,0,0,Between -11 and 5
+99,Melatonin,232.28,54.1,270.0,0.8,17,2,2,4,232.121,232.121,0,1,0,0,0,0,0,0,0,Between -11 and 5
+100,Carbamazepine,236.27,46.3,326.0,2.5,18,1,1,0,236.095,236.095,0,1,0,0,0,0,0,0,0,Between -11 and 5
+101,Clofazimine,473.4,40.0,829.0,7.1,33,1,4,4,472.122,472.122,0,1,0,0,0,0,0,0,0,Larger than 6
+102,Diphenhydramine,255.35,12.5,211.0,3.3,19,0,2,6,255.162,255.162,0,1,0,0,0,0,0,0,0,Between -11 and 5
+103,Thalidomide,258.23,83.6,449.0,0.3,19,1,4,1,258.064,258.064,0,1,0,1,0,1,0,0,0,Between -11 and 5
+104,Hydrocortisone,362.5,94.8,684.0,1.6,26,3,5,2,362.209,362.209,0,1,0,7,7,0,0,0,0,Between -11 and 5
+105,Progesterone,314.5,34.1,589.0,3.9,23,0,2,1,314.225,314.225,0,1,0,6,6,0,0,0,0,Between -11 and 5
+106,o-Aminoazotoluene,225.29,50.7,264.0,3.7,17,1,3,2,225.127,225.127,0,1,0,0,0,0,0,0,0,Between -11 and 5
+107,Clonitralid,388.2,141.0,414.0,,25,4,6,3,387.039,387.039,0,2,0,0,0,0,0,0,0,Larger than 6
+108,Pimozide,461.5,35.6,632.0,6.3,34,1,4,7,461.228,461.228,0,1,0,0,0,0,0,0,0,Larger than 6
+109,Nitazoxanide,307.28,142.0,428.0,2.0,21,1,7,4,307.026,307.026,0,1,0,0,0,0,0,0,0,Between -11 and 5
+110,Deferoxamine hydrochloride,597.1,206.0,739.0,,40,7,9,23,596.33,596.33,0,2,0,0,0,0,0,0,0,Larger than 6
+111,Chloroquine monophosphate,417.9,106.0,359.0,,27,4,7,8,417.158,417.158,0,2,0,1,0,1,0,0,0,Larger than 6
+112,Retinol,286.5,20.2,496.0,5.7,21,1,1,5,286.23,286.23,0,1,0,0,0,0,4,4,0,Larger than 6
+113,Melphalan,305.2,66.6,265.0,-0.5,19,2,4,8,304.075,304.075,0,1,0,1,1,0,0,0,0,Between -11 and 5
+114,Dasatinib,488.0,135.0,642.0,3.6,33,3,9,7,487.156,487.156,0,1,0,0,0,0,0,0,0,Between -11 and 5
+115,Masitinib,498.6,102.0,696.0,4.3,36,2,7,7,498.22,498.22,0,1,0,0,0,0,0,0,0,Between -11 and 5
+116,Acrivastine and pseudoephedrine hydrochloride,550.1,85.7,635.0,,39,4,6,9,549.276,549.276,0,3,0,2,2,0,2,2,0,Larger than 6
+117,"4-(carboxymethyl)-2-((R)-1-(2-(2,5-dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-1,3,2-dioxaborinane-4-carboxylic acid",517.1,168.0,815.0,,34,4,9,10,516.087,516.087,0,1,0,2,1,1,0,0,0,Larger than 6
+118,Remdesivir,602.6,204.0,1010.0,1.9,42,4,13,14,602.225,602.225,0,1,0,6,6,0,0,0,0,Between -11 and 5
+119,"Ivermectin B1a, epi-",875.1,170.0,1680.0,4.1,62,3,14,8,874.508,874.508,0,1,0,20,20,0,3,3,0,Between -11 and 5
+120,Hydroxocobalamin,1270.4,452.0,3140.0,-3.4,90,9,19,26,1269.63,1269.63,-2,1,0,14,0,14,3,3,0,Between -11 and 5
+121,Zanubrutinib,471.5,103.0,756.0,3.5,35,2,5,6,471.227,471.227,0,1,0,1,1,0,0,0,0,Between -11 and 5
+122,Gamma-Aminobutyric Acid,103.12,63.3,62.7,-3.2,7,2,3,3,103.063,103.063,0,1,0,0,0,0,0,0,0,Between -11 and 5
+123,Caffeine,194.19,58.4,293.0,-0.1,14,0,3,0,194.08,194.08,0,1,0,0,0,0,0,0,0,Between -11 and 5
+124,Dapsone,248.3,94.6,306.0,1.0,17,2,4,2,248.062,248.062,0,1,0,0,0,0,0,0,0,Between -11 and 5
+125,Leflunomide,270.21,55.1,327.0,2.5,19,1,6,2,270.062,270.062,0,1,0,0,0,0,0,0,0,Between -11 and 5
+126,Raloxifene,473.6,98.2,655.0,6.1,34,2,6,7,473.166,473.166,0,1,0,0,0,0,0,0,0,Larger than 6
+127,Imatinib,493.6,86.3,706.0,3.5,37,2,7,7,493.259,493.259,0,1,0,0,0,0,0,0,0,Between -11 and 5
+128,Dexamethasone,392.5,94.8,805.0,1.9,28,3,6,2,392.2,392.2,0,1,0,8,8,0,0,0,0,Between -11 and 5
+129,Colchicine,399.4,83.1,740.0,1.0,29,1,6,5,399.168,399.168,0,1,0,1,1,0,0,0,0,Between -11 and 5
+130,Estradiol cypionate,396.6,46.5,597.0,7.1,29,1,3,5,396.266,396.266,0,1,0,5,5,0,0,0,0,Larger than 6
+131,Cetylpyridinium Chloride,340.0,3.9,208.0,,23,0,1,15,339.269,339.269,0,2,0,0,0,0,0,0,0,Larger than 6
+132,Plerixafor,502.8,78.7,456.0,0.0,36,6,8,4,502.447,502.447,0,1,0,0,0,0,0,0,0,Between -11 and 5
+133,Telmisartan,514.6,72.9,831.0,6.9,39,1,4,7,514.237,514.237,0,1,0,0,0,0,0,0,0,Larger than 6
+134,Sorafenib,464.8,92.4,646.0,4.1,32,3,7,5,464.086,464.086,0,1,0,0,0,0,0,0,0,Between -11 and 5
+135,Bacitracin zinc,1488.1,552.0,2950.0,,101,16,21,31,1485.68,1485.68,0,2,0,16,0,16,0,0,0,Larger than 6
+136,Hymecromone,176.17,46.5,257.0,1.9,13,1,3,0,176.047,176.047,0,1,0,0,0,0,0,0,0,Between -11 and 5
+137,Cob(II)alamin,1329.3,452.0,3150.0,,91,9,19,16,1328.56,1328.56,-2,2,0,14,14,0,3,3,0,Larger than 6
+138,Tofacitinib,312.37,88.9,488.0,1.5,23,1,5,3,312.17,312.17,0,1,0,2,2,0,0,0,0,Between -11 and 5
+139,Elsulfavirine,629.3,134.0,977.0,5.2,37,2,7,8,626.943,626.943,0,1,0,0,0,0,0,0,0,Larger than 6
+140,Fostamatinib,580.5,187.0,904.0,1.6,40,4,15,10,580.148,580.148,0,1,0,0,0,0,0,0,0,Between -11 and 5
+141,3-(4-chlorophenyl)-N-(pyridin-4-ylmethyl)adamantane-1-carboxamide,380.9,42.0,551.0,4.7,27,1,2,4,380.166,380.166,0,1,0,2,0,2,0,0,0,Between -11 and 5
+142,Adenosylcobalamin,1579.6,571.0,3730.0,,109,12,27,17,1578.66,1578.66,-3,3,0,18,18,0,3,3,0,Larger than 6
+143,Solnatide,1923.1,861.0,4170.0,-10.7,134,27,32,27,1921.81,1921.81,0,1,0,16,16,0,0,0,0,Smaller than -10
+144,Subasumstat,578.1,193.0,942.0,3.5,38,4,11,8,577.122,577.122,0,1,0,4,4,0,0,0,0,Between -11 and 5
+145,Lactoferrin,3125.8,1360.0,7330.0,6.8,219,51,65,108,3124.68,3123.68,0,1,0,26,26,0,0,0,0,Larger than 6
+146,Compstatin 40,1789.1,692.0,3770.0,-2.1,126,21,23,30,1787.84,1787.84,0,1,0,15,15,0,0,0,0,Between -11 and 5
+147,Celecoxib,381.4,86.4,577.0,3.4,26,1,7,3,381.076,381.076,0,1,0,0,0,0,0,0,0,Between -11 and 5
+148,Eucalyptol,154.25,9.2,164.0,2.5,11,0,1,0,154.136,154.136,0,1,0,0,0,0,0,0,0,Between -11 and 5
+149,Disulfiram,296.5,121.0,201.0,3.9,16,0,4,7,296.051,296.051,0,1,0,0,0,0,0,0,0,Between -11 and 5
+150,Ibuprofen,206.28,37.3,203.0,3.5,15,1,2,4,206.131,206.131,0,1,0,1,0,1,0,0,0,Between -11 and 5
+151,Niclosamide,327.12,95.2,404.0,4.0,21,2,4,2,325.986,325.986,0,1,0,0,0,0,0,0,0,Between -11 and 5
+152,Simvastatin,418.6,72.8,706.0,4.7,30,1,5,7,418.272,418.272,0,1,0,7,7,0,0,0,0,Between -11 and 5
+153,Methotrexate,454.4,211.0,704.0,-1.8,33,5,12,9,454.171,454.171,0,1,0,1,1,0,0,0,0,Between -11 and 5
+154,Angiotensin II,1046.2,409.0,1980.0,-1.7,75,13,15,29,1045.53,1045.53,0,1,0,9,9,0,0,0,0,Between -11 and 5
+155,Lenalidomide,259.26,92.5,437.0,-0.5,19,2,4,1,259.096,259.096,0,1,0,1,0,1,0,0,0,Between -11 and 5
+156,Aspartyl-alanyl-diketopiperazine,186.17,95.5,263.0,-1.4,13,3,4,2,186.064,186.064,0,1,0,2,2,0,0,0,0,Between -11 and 5
+157,Silmitasertib,349.8,75.1,491.0,4.4,25,2,5,3,349.062,349.062,0,1,0,0,0,0,0,0,0,Between -11 and 5
+158,Ibrutinib,440.5,99.2,678.0,3.6,33,1,6,5,440.196,440.196,0,1,0,1,1,0,0,0,0,Between -11 and 5
+159,Bemcentinib,506.6,97.8,775.0,5.5,38,2,7,4,506.291,506.291,0,1,0,1,1,0,0,0,0,Larger than 6
+160,Teriflunomide,270.21,73.1,426.0,3.3,19,2,6,2,270.062,270.062,0,1,0,0,0,0,1,1,0,Between -11 and 5
+161,Tazemetostat,572.7,83.1,992.0,4.2,42,2,6,9,572.336,572.336,0,1,0,0,0,0,0,0,0,Between -11 and 5
+162,Acalabrutinib,465.5,119.0,845.0,3.0,35,2,6,4,465.191,465.191,0,1,0,1,1,0,0,0,0,Between -11 and 5
+163,Ifenprodil,325.4,43.7,353.0,3.9,24,2,3,5,325.204,325.204,0,1,0,2,0,2,0,0,0,Between -11 and 5
+164,Ursodeoxycholic acid,392.6,77.8,605.0,4.9,28,3,4,4,392.293,392.293,0,1,0,10,10,0,0,0,0,Between -11 and 5
+165,Metoprolol succinate,652.8,176.0,308.0,,46,6,12,21,652.393,652.393,0,3,0,2,0,2,0,0,0,Larger than 6
+166,Merimepodib,452.5,124.0,652.0,2.1,33,3,7,8,452.17,452.17,0,1,0,1,1,0,0,0,0,Between -11 and 5
+167,Fludrocortisone acetate,422.5,101.0,838.0,1.7,30,2,7,4,422.21,422.21,0,1,0,7,7,0,0,0,0,Between -11 and 5
+168,Triazavirin,228.19,141.0,435.0,0.4,15,1,6,1,228.007,228.007,0,1,0,0,0,0,0,0,0,Between -11 and 5
+169,Ceftriaxone sodium,598.6,297.0,1120.0,,38,2,14,7,598.01,598.01,0,3,0,2,2,0,1,1,0,Larger than 6
+170,Rivaroxaban,435.9,116.0,645.0,2.5,29,1,6,5,435.066,435.066,0,1,0,1,1,0,0,0,0,Between -11 and 5
+171,Ventolin,337.39,156.0,309.0,,22,6,8,5,337.12,337.12,0,2,0,1,0,1,0,0,0,Larger than 6
+172,Apixaban,459.5,111.0,777.0,2.2,34,1,5,5,459.191,459.191,0,1,0,0,0,0,0,0,0,Between -11 and 5
+173,Ceftriaxone disodium salt hemiheptahydrate,1323.2,601.0,1120.0,,83,11,35,14,1322.09,1322.09,0,13,0,4,4,0,2,2,0,Larger than 6
+174,Vadadustat,306.7,99.5,393.0,2.5,21,3,5,4,306.041,306.041,0,1,0,0,0,0,0,0,0,Between -11 and 5
+175,Carbohydrate moiety of bromelain,1026.9,483.0,1680.0,-11.6,70,18,29,16,1026.38,1026.38,0,1,0,29,28,1,0,0,0,Smaller than -10
+176,Siponimod fumarate,1149.3,199.0,896.0,,82,4,20,20,1148.53,1148.53,0,3,0,0,0,0,3,3,0,Larger than 6
+177,(s)-1-(3-Chloro-4-fluorophenyl)ethanamine hydrochloride,210.07,26.0,131.0,,12,2,2,1,209.017,209.017,0,2,0,1,1,0,0,0,0,Larger than 6
+178,Prezcobix,1323.7,343.0,1980.0,,92,6,19,32,1322.59,1322.59,0,2,0,8,8,0,0,0,0,Larger than 6
+179,"[(2R,3R,4R,5R)-5-(4-amino-5-deuteriopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-bis(2-methylpropanoyloxy)oxolan-2-yl]methyl 2-methylpropanoate;hydrobromide",583.4,168.0,887.0,,37,2,11,11,582.155,582.155,0,2,1,4,4,0,0,0,0,Larger than 6
+180,Berberine,336.4,40.8,488.0,3.6,25,0,4,2,336.124,336.124,1,1,0,0,0,0,0,0,0,Between -11 and 5
+181,Cyproheptadine,287.4,3.2,423.0,4.7,22,0,1,0,287.167,287.167,0,1,0,0,0,0,0,0,0,Between -11 and 5
+182,Doxazosin,451.5,112.0,678.0,2.5,33,1,9,4,451.186,451.186,0,1,0,1,0,1,0,0,0,Between -11 and 5
+183,Fluconazole,306.27,81.6,358.0,0.4,22,1,7,5,306.104,306.104,0,1,0,0,0,0,0,0,0,Between -11 and 5
+184,Irbesartan,428.5,87.1,682.0,4.1,32,1,5,7,428.232,428.232,0,1,0,0,0,0,0,0,0,Between -11 and 5
+185,Sulfamethoxazole,253.28,107.0,346.0,0.9,17,2,6,3,253.052,253.052,0,1,0,0,0,0,0,0,0,Between -11 and 5
+186,D-Glucose,180.16,110.0,151.0,-2.6,12,5,6,1,180.063,180.063,0,1,0,5,4,1,0,0,0,Between -11 and 5
+187,N-Vinyl-2-pyrrolidone,111.14,20.3,120.0,0.4,8,0,1,1,111.068,111.068,0,1,0,0,0,0,0,0,0,Between -11 and 5
+188,Trimetazidine,266.34,43.0,259.0,1.0,19,1,5,5,266.163,266.163,0,1,0,0,0,0,0,0,0,Between -11 and 5
+189,Estetrol,304.4,80.9,441.0,1.5,22,4,4,0,304.167,304.167,0,1,0,7,7,0,0,0,0,Between -11 and 5
+190,Deferoxamine mesylate,656.8,269.0,832.0,,44,7,12,23,656.341,656.341,0,2,0,0,0,0,0,0,0,Larger than 6
+191,Tramadol Hydrochloride,299.83,32.7,282.0,,20,2,3,4,299.165,299.165,0,2,0,2,2,0,0,0,0,Larger than 6
+192,Nebivolol,405.4,71.0,483.0,3.0,29,3,7,6,405.175,405.175,0,1,0,4,0,4,0,0,0,Between -11 and 5
+193,Argatroban monohydrate,526.7,190.0,887.0,,36,6,9,9,526.257,526.257,0,2,0,4,3,1,0,0,0,Larger than 6
+194,Sulfamethoxazole and trimethoprim,543.6,212.0,653.0,,38,4,13,8,543.19,543.19,0,2,0,0,0,0,0,0,0,Larger than 6
+195,Zinc Gluconate,455.7,283.0,165.0,,27,10,14,8,454.03,454.03,0,3,0,8,8,0,0,0,0,Larger than 6
+196,"Zinc;(2R,3S,4R,5R)-2,3,4,5,6-pentahydroxyhexanoate",455.7,283.0,385.0,,27,10,14,8,454.03,454.03,0,3,0,8,8,0,0,0,0,Larger than 6
+197,(+)-Mefloquine,378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,2,0,0,0,0,Between -11 and 5
+198,"(2S,5R,6R)-6-[[(2S)-2-[(4-ethyl-2,3-dioxo-piperazine-1-carbonyl)amino]-2-phenyl-propanoyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid; (2S,3S,5R)-3-methyl-4,4,7-trioxo-3-(triazol-1-ylmethyl)-4$l^{6}-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid",831.9,313.0,1600.0,,57,4,15,9,831.232,831.232,0,2,0,7,7,0,0,0,0,Larger than 6
+199,Favipiravir,157.1,84.6,282.0,-0.6,11,2,4,1,157.029,157.029,0,1,0,0,0,0,0,0,0,Between -11 and 5
+200,Maraviroc,513.7,63.0,751.0,5.1,37,1,6,8,513.328,513.328,0,1,0,3,3,0,0,0,0,Larger than 6
+201,Toremifene,406.0,12.5,483.0,7.2,29,0,2,9,405.186,405.186,0,1,0,0,0,0,1,1,0,Larger than 6
+202,"(3-methyl-2,4-dioxo-3,4-dihydroquinazolin-1(2H)-yl)acetic acid",234.21,77.9,369.0,0.4,17,1,4,2,234.064,234.064,0,1,0,0,0,0,0,0,0,Between -11 and 5
+203,Enalapril maleate,492.5,171.0,638.0,,35,4,10,12,492.211,492.211,0,2,0,3,3,0,1,1,0,Larger than 6
+204,Naltrexone hydrochloride,377.9,70.0,621.0,,26,3,5,2,377.139,377.139,0,2,0,4,4,0,0,0,0,Larger than 6
+205,Chlorhexidine Gluconate,897.8,455.0,819.0,,60,18,16,23,896.32,896.32,0,3,0,8,8,0,2,2,0,Larger than 6
+206,Piperacillin/tazobactam,817.9,313.0,1550.0,,56,4,15,9,817.216,817.216,0,2,0,7,7,0,0,0,0,Larger than 6
+207,3-[5-(azetidine-1-carbonyl)pyrazin-2-yl]oxy-5-[(2S)-1-methoxypropan-2-yl]oxy-N-(5-methylpyrazin-2-yl)benzamide,478.5,129.0,710.0,1.3,35,1,9,9,478.196,478.196,0,1,0,1,1,0,0,0,0,Between -11 and 5
+208,Degarelix,1632.3,513.0,3390.0,3.5,117,17,18,41,1630.75,1630.75,0,1,0,11,11,0,0,0,0,Between -11 and 5
+209,(3S)-3-amino-6-(diaminomethylideneazaniumyl)hex-1-en-2-olate,172.23,115.0,174.0,-0.8,12,4,2,5,172.132,172.132,0,1,0,1,1,0,0,0,0,Between -11 and 5
+210,Sivelestat sodium,528.5,154.0,738.0,,35,6,12,9,528.139,528.139,0,6,0,0,0,0,0,0,0,Larger than 6
+211,CID 23679441,576.6,291.0,1120.0,,37,3,13,8,576.028,576.028,0,2,0,2,2,0,1,1,0,Larger than 6
+212,Masitinib mesylate,594.8,164.0,788.0,,41,3,10,7,594.208,594.208,0,2,0,0,0,0,0,0,0,Larger than 6
+213,"[(1S,4R,6S,7Z,18R)-4-(cyclopropylsulfonylcarbamoyl)-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-3,16-diazatricyclo[14.3.0.04,6]nonadec-7-en-18-yl] 4-fluoro-1,3-dihydroisoindole-2-carboxylate",731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,4,1,1,1,0,Between -11 and 5
+214,Natrii chloridi solutio composita,309.11,63.4,24.8,,13,4,8,0,307.852,307.852,0,10,0,0,0,0,0,0,0,Larger than 6
+215,CID 87060529,577.6,288.0,1110.0,,37,4,13,8,577.036,577.036,0,2,0,2,2,0,1,1,0,Larger than 6
+216,"(2S,3R)-3-[(2-aminopyridin-4-yl)methyl]-1-[[(1R)-1-cyclooctylethyl]carbamoyl]-4-oxoazetidine-2-carboxylic acid",402.5,126.0,606.0,3.7,29,3,6,5,402.227,402.227,0,1,0,3,3,0,0,0,0,Between -11 and 5
+217,Sinapultide acetate,2529.5,813.0,4910.0,,178,28,30,93,2528.85,2527.85,0,2,0,21,21,0,0,0,0,Larger than 6
+218,Bemnifosbuvir hemisulfate,1261.1,453.0,1000.0,,85,10,32,24,1260.4,1260.4,0,3,0,12,12,0,0,0,0,Larger than 6
+219,Reamberin,357.27,193.0,216.0,,23,6,10,7,357.101,357.101,0,4,0,4,4,0,0,0,0,Larger than 6
+220,Pomotrelvir,455.9,127.0,779.0,3.1,32,4,4,8,455.172,455.172,0,1,0,3,3,0,0,0,0,Between -11 and 5
+221,Selenious acid,128.99,57.5,26.3,,4,2,3,0,129.917,129.917,0,1,0,0,0,0,0,0,0,Larger than 6
+222,Spermidine,145.25,64.099,56.8,-1.0,10,3,3,7,145.158,145.158,0,1,0,0,0,0,0,0,0,Between -11 and 5
+223,Salbutamol,239.31,72.7,227.0,0.3,17,4,4,5,239.152,239.152,0,1,0,1,0,1,0,0,0,Between -11 and 5
+224,Bromhexine,376.13,29.3,256.0,4.3,18,1,2,3,375.997,373.999,0,1,0,0,0,0,0,0,0,Between -11 and 5
+225,Ebselen,274.19,20.3,275.0,,16,0,1,1,274.985,274.985,0,1,0,0,0,0,0,0,0,Larger than 6
+226,Fluoxetine,309.33,21.3,308.0,4.0,22,1,5,6,309.134,309.134,0,1,0,1,0,1,0,0,0,Between -11 and 5
+227,Ketotifen,309.4,48.6,476.0,3.2,22,0,3,0,309.119,309.119,0,1,0,0,0,0,0,0,0,Between -11 and 5
+228,Mefloquine,378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,0,2,0,0,0,Between -11 and 5
+229,Midazolam,325.8,30.2,471.0,2.5,23,0,3,1,325.078,325.078,0,1,0,0,0,0,0,0,0,Between -11 and 5
+230,Nitroglycerin,227.09,165.0,219.0,1.6,15,0,9,5,227.003,227.003,0,1,0,0,0,0,0,0,0,Between -11 and 5
+231,Quetiapine,383.5,73.6,496.0,2.1,27,1,5,6,383.167,383.167,0,1,0,0,0,0,0,0,0,Between -11 and 5
+232,Liothyronine,650.97,92.8,402.0,1.7,23,3,5,5,650.79,650.79,0,1,0,1,1,0,0,0,0,Between -11 and 5
+233,Pyridostigmine bromide,261.12,33.4,183.0,,14,0,3,2,260.016,260.016,0,2,0,0,0,0,0,0,0,Larger than 6
+234,Psilocybine,284.25,85.8,347.0,-1.6,19,3,5,5,284.093,284.093,0,1,0,0,0,0,0,0,0,Between -11 and 5
+235,Luminol,177.16,84.2,254.0,0.3,13,3,3,0,177.054,177.054,0,1,0,0,0,0,0,0,0,Between -11 and 5
+236,Acetylcysteine,163.2,67.4,148.0,0.4,10,3,4,3,163.03,163.03,0,1,0,1,1,0,0,0,0,Between -11 and 5
+237,Cyproheptadine hydrochloride,323.9,3.2,423.0,,23,1,1,0,323.144,323.144,0,2,0,0,0,0,0,0,0,Larger than 6
+238,Carmoisine,502.4,176.0,854.0,,33,1,9,2,501.988,501.988,0,3,0,0,0,0,0,0,0,Larger than 6
+239,Etoposide,588.6,161.0,969.0,0.6,42,3,13,5,588.184,588.184,0,1,0,10,10,0,0,0,0,Between -11 and 5
+240,Albuterol Sulfate,576.7,228.0,309.0,,39,10,12,10,576.272,576.272,0,3,0,2,0,2,0,0,0,Larger than 6
+241,Dexbudesonide,430.5,93.1,862.0,2.5,31,2,6,4,430.236,430.236,0,1,0,9,9,0,0,0,0,Between -11 and 5
+242,"2,2-Dimethyl-4-(chloromethyl)-1,3-dioxa-2-silacyclopentane",166.68,18.5,107.0,,9,0,2,1,166.022,166.022,0,1,0,1,0,1,0,0,0,Larger than 6
+243,Clopidogrel,321.8,57.8,381.0,3.8,21,0,4,4,321.059,321.059,0,1,0,1,1,0,0,0,0,Between -11 and 5
+244,Valsartan,435.5,112.0,608.0,4.4,32,2,6,10,435.227,435.227,0,1,0,1,1,0,0,0,0,Between -11 and 5
+245,Tirofiban,440.6,113.0,579.0,1.4,30,3,7,14,440.234,440.234,0,1,0,1,1,0,0,0,0,Between -11 and 5
+246,Voriconazole,349.31,76.7,448.0,1.5,25,1,8,5,349.115,349.115,0,1,0,2,2,0,0,0,0,Between -11 and 5
+247,N-Phenylethylenediamine,136.19,38.0,77.3,0.6,10,2,2,3,136.1,136.1,0,1,0,0,0,0,0,0,0,Between -11 and 5
+248,Oseltamivir phosphate,410.4,168.0,468.0,,27,5,9,8,410.182,410.182,0,2,0,3,3,0,0,0,0,Larger than 6
+249,Nicotine,162.23,16.1,147.0,1.2,12,0,2,1,162.116,162.116,0,1,0,1,1,0,0,0,0,Between -11 and 5
+250,Lactose monohydrate,360.31,191.0,382.0,,24,9,12,4,360.127,360.127,0,2,0,10,10,0,0,0,0,Larger than 6
+251,"2,4-Dioxaspiro[5.5]undec-8-ene, 3-(2-furanyl)-",220.26,31.6,266.0,2.1,16,0,3,1,220.11,220.11,0,1,0,0,0,0,0,0,0,Between -11 and 5
+252,Sivelestat,434.5,147.0,731.0,3.0,30,3,8,9,434.115,434.115,0,1,0,0,0,0,0,0,0,Between -11 and 5
+253,Tafenoquine,463.5,78.6,597.0,5.4,33,2,9,9,463.208,463.208,0,1,0,1,0,1,0,0,0,Larger than 6
+254,IB-Meca,510.3,134.0,589.0,0.9,29,4,8,5,510.051,510.051,0,1,0,4,4,0,0,0,0,Between -11 and 5
+255,"(S)-Hexahydropyrrolo[1,2-a]pyrazine-1,4-dione",154.17,49.4,215.0,-0.6,11,1,2,0,154.074,154.074,0,1,0,1,1,0,0,0,0,Between -11 and 5
+256,Arbidol,477.4,80.0,546.0,4.4,29,1,5,8,476.077,476.077,0,1,0,0,0,0,0,0,0,Between -11 and 5
+257,Tempol,172.24,24.5,159.0,0.9,12,1,2,0,172.134,172.134,0,1,0,0,0,0,0,0,0,Between -11 and 5
+258,Atazanavir,704.9,171.0,1110.0,5.6,51,5,9,18,704.39,704.39,0,1,0,4,4,0,0,0,0,Larger than 6
+259,Centhaquine,331.5,19.4,404.0,4.5,25,0,3,4,331.205,331.205,0,1,0,0,0,0,0,0,0,Between -11 and 5
+260,Regadenoson,390.35,187.0,587.0,-1.5,28,5,10,4,390.14,390.14,0,1,0,4,4,0,0,0,0,Between -11 and 5
+261,Teprotide,1101.3,387.0,2330.0,-0.9,79,10,13,24,1100.58,1100.58,0,1,0,10,10,0,0,0,0,Between -11 and 5
+262,Azithromycin,749.0,180.0,1150.0,4.0,52,5,14,7,748.509,748.509,0,1,0,18,18,0,0,0,0,Between -11 and 5
+263,Posaconazole,700.8,112.0,1170.0,4.6,51,1,11,12,700.33,700.33,0,1,0,4,4,0,0,0,0,Between -11 and 5
+264,Cannabidiol,314.5,40.5,414.0,6.5,23,2,2,6,314.225,314.225,0,1,0,2,2,0,0,0,0,Larger than 6
+265,"(R)-[2,8-bis(trifluoromethyl)-4-quinolyl]-[(2R)-2-piperidyl]methanol",378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,2,0,0,0,0,Between -11 and 5
+266,Tetramethylol-melamin-dioxy-propylen [German],358.35,170.0,377.0,-0.8,25,6,12,11,358.16,358.16,0,1,0,2,0,2,0,0,0,Between -11 and 5
+267,Heme arginate,792.7,260.0,1180.0,,55,8,10,13,792.305,792.305,2,3,0,1,1,0,0,0,0,Larger than 6
+268,Zincacetate,185.5,74.6,78.6,,9,2,4,0,183.971,183.971,0,3,0,0,0,0,0,0,0,Larger than 6
+269,Montelukast,586.2,95.7,891.0,7.7,41,2,5,12,585.21,585.21,0,1,0,1,1,0,1,1,0,Larger than 6
+270,Crocetin,328.4,74.6,608.0,5.4,24,2,4,8,328.167,328.167,0,1,0,0,0,0,7,7,0,Larger than 6
+271,Fondaparinux,1508.3,873.0,3450.0,-14.7,91,19,52,30,1506.95,1506.95,0,1,0,25,25,0,0,0,0,Smaller than -10
+272,Fluvoxamine,318.33,56.8,327.0,2.6,22,1,7,9,318.156,318.156,0,1,0,0,0,0,1,1,0,Between -11 and 5
+273,Naltrexone,341.4,70.0,621.0,1.9,25,2,5,2,341.163,341.163,0,1,0,4,4,0,0,0,0,Between -11 and 5
+274,20-Hydroxyecdysone,480.6,138.0,869.0,0.5,34,6,7,5,480.309,480.309,0,1,0,10,10,0,0,0,0,Between -11 and 5
+275,Ceftriaxone,554.6,288.0,1110.0,-1.3,36,4,13,8,554.046,554.046,0,1,0,2,2,0,1,1,0,Between -11 and 5
+276,"(3E,4S)-4-Hydroxy-3-{2-[(1R,4aS,5R,6R,8aS)-6-hydroxy-5-(hydroxymethyl)-5,8a-dimethyl-2-methylenedecahydronaphthalen-1-yl]ethylidene}dihydrofuran-2(3H)-one",350.4,87.0,597.0,2.2,25,3,5,3,350.209,350.209,0,1,0,6,2,4,1,1,0,Between -11 and 5
+277,Artemether and lumefantrine,827.3,69.6,1100.0,,56,1,7,11,825.333,825.333,0,2,0,9,7,2,1,1,0,Larger than 6
+278,Isavuconazonium,717.8,188.0,1210.0,4.1,51,2,13,15,717.242,717.242,1,1,0,3,2,1,0,0,0,Between -11 and 5
+279,Luminol sodium salt,200.15,84.2,254.0,,14,3,3,0,200.044,200.044,0,2,0,0,0,0,0,0,0,Larger than 6
+280,Dapagliflozin,408.9,99.4,472.0,2.3,28,4,6,6,408.134,408.134,0,1,0,5,5,0,0,0,0,Between -11 and 5
+281,Zinc Picolinate,309.6,106.0,108.0,,19,0,6,0,307.978,307.978,0,3,0,0,0,0,0,0,0,Larger than 6
+282,"Pregna-1,4-diene-3,20-dione,21-(3-carboxy-1-oxopropoxy)-11,17-dihydroxy-6-methyl-, monosodiumsalt, (6a,11b)-",497.5,138.0,981.0,,35,3,8,7,497.215,497.215,0,2,0,8,8,0,0,0,0,Larger than 6
+283,Vortioxetine,298.4,40.6,316.0,4.2,21,1,3,3,298.15,298.15,0,1,0,0,0,0,0,0,0,Between -11 and 5
+284,Enisamium iodide,354.19,33.0,241.0,,18,1,2,3,354.023,354.023,0,2,0,0,0,0,0,0,0,Larger than 6
+285,Cenicriviroc,696.9,105.0,1060.0,7.5,50,1,7,17,696.371,696.371,0,1,0,1,1,0,1,1,0,Larger than 6
+286,Apremilast,460.5,128.0,825.0,1.8,32,1,7,8,460.13,460.13,0,1,0,1,1,0,0,0,0,Between -11 and 5
+287,Empagliflozin,450.9,109.0,558.0,2.0,31,4,7,6,450.145,450.145,0,1,0,6,6,0,0,0,0,Between -11 and 5
+288,N6-ethanimidoyl-D-lysine,187.24,102.0,192.0,-3.1,13,3,4,6,187.132,187.132,0,1,0,1,1,0,0,0,0,Between -11 and 5
+289,Emricasan,569.5,151.0,934.0,3.6,40,4,11,11,569.179,569.179,0,1,0,2,2,0,0,0,0,Between -11 and 5
+290,Aviptadil Acetate,3344.9,1480.0,7510.0,-13.7,234,51,51,116,3343.74,3342.73,0,1,0,31,0,31,0,0,0,Smaller than -10
+291,Quinine sulfate dihydrate,782.9,176.0,538.0,,55,6,14,8,782.356,782.356,0,5,0,8,8,0,0,0,0,Larger than 6
+292,Hydrocortisone 21-hemisuccinate sodium salt,485.5,138.0,908.0,,34,3,8,7,485.215,485.215,0,2,0,7,7,0,0,0,0,Larger than 6
+293,Liothyronine sodium,672.95,95.6,408.0,,24,2,5,5,672.772,672.772,0,2,0,1,1,0,0,0,0,Larger than 6
+294,sodium;8-amino-4-oxo-3H-phthalazin-1-olate,199.14,90.5,269.0,,14,2,4,0,199.036,199.036,0,2,0,0,0,0,0,0,0,Larger than 6
+295,Edoxaban tosylate monohydrate,738.3,229.0,1090.0,,49,5,12,6,737.207,737.207,0,3,0,3,3,0,0,0,0,Larger than 6
+296,Daclatasvir,738.9,175.0,1190.0,5.1,54,4,8,13,738.385,738.385,0,1,0,4,4,0,0,0,0,Larger than 6
+297,"2-[(4S)-4-amino-5,5-dihydroxyhexyl]guanidine",190.24,131.0,177.0,-2.5,13,5,4,5,190.143,190.143,0,1,0,1,1,0,0,0,0,Between -11 and 5
+298,Fosmanogepix,468.4,148.0,644.0,1.6,33,2,9,9,468.12,468.12,0,1,0,0,0,0,0,0,0,Between -11 and 5
+299,Larazotide acetate,785.9,339.0,1320.0,,55,10,13,21,785.428,785.428,0,2,0,5,5,0,0,0,0,Larger than 6
+300,Thymosin,3051.3,1370.0,6930.0,-22.0,212,47,57,109,3050.5,3049.5,0,1,0,30,30,0,0,0,0,Smaller than -10
+301,Sofosbuvir,529.5,153.0,913.0,1.0,36,3,11,11,529.163,529.163,0,1,0,6,6,0,0,0,0,Between -11 and 5
+302,Razuprotafib,586.7,212.0,906.0,3.7,39,4,10,12,586.101,586.101,0,1,0,2,2,0,0,0,0,Between -11 and 5
+303,Nalpha-[(4-Methylpiperazin-1-Yl)carbonyl]-N-[(3s)-1-Phenyl-5-(Phenylsulfonyl)pentan-3-Yl]-L-Phenylalaninamide,576.8,107.0,897.0,4.4,41,2,5,12,576.277,576.277,0,1,0,2,2,0,0,0,0,Between -11 and 5
+304,Ascorbic Acid,176.12,107.0,232.0,-1.6,12,4,6,2,176.032,176.032,0,1,0,2,2,0,0,0,0,Between -11 and 5
+305,Doxycycline,444.4,182.0,956.0,-0.7,32,6,9,2,444.153,444.153,0,1,0,6,6,0,0,0,0,Between -11 and 5
+306,Rabeximod,409.9,63.0,590.0,3.9,29,1,4,5,409.167,409.167,0,1,0,0,0,0,0,0,0,Between -11 and 5
+307,Kaolin,258.16,98.0,167.0,,13,2,9,4,257.902,257.902,0,3,0,0,0,0,0,0,0,Larger than 6
+308,H-Ile-OH.H-Thr-OH.H-Leu-OH.H-Val-OH.H-Met-OH.H-Phe-OH.H-Trp-OH.H-Lys-OH,1163.4,594.0,988.0,,80,19,27,25,1162.65,1162.65,0,8,0,10,10,0,0,0,0,Larger than 6
+309,Semaglutide,4114.0,1650.0,9590.0,-5.8,291,57,63,151,4112.12,4111.12,0,1,0,30,30,0,0,0,0,Between -11 and 5
+310,Isuzinaxib hydrochloride,315.8,45.2,412.0,,22,2,3,4,315.114,315.114,0,2,0,0,0,0,0,0,0,Larger than 6
+311,CID 66726979,457.5,147.0,731.0,,31,3,8,9,457.105,457.105,0,2,0,0,0,0,0,0,0,Larger than 6
+312,Desidustat,332.31,116.0,583.0,1.9,24,3,6,6,332.101,332.101,0,1,0,0,0,0,0,0,0,Between -11 and 5
+313,"7-[[2-Ethoxyimino-2-[5-(phosphonoamino)-1,2,4-thiadiazol-3-yl]acetyl]amino]-3-[[4-(1-methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid;acetate",744.7,368.0,1240.0,,47,5,19,11,744.031,744.031,0,2,0,2,0,2,1,0,1,Larger than 6
+314,1-(4-(((6-Amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)-4-fluoropiperidin-1-yl)prop-2-en-1-one,447.5,93.4,643.0,4.1,33,2,7,7,447.207,447.207,0,1,0,0,0,0,0,0,0,Between -11 and 5
+315,Ceftriaxone disodium hemiheptahydrate,600.6,288.0,1110.0,,38,4,13,8,600.026,600.026,0,3,0,2,2,0,1,1,0,Larger than 6
+316,Descovy,723.7,257.0,1050.0,,49,4,15,14,723.236,723.236,0,2,0,5,5,0,0,0,0,Larger than 6
+317,Zimlovisertib,361.4,104.0,535.0,2.0,26,2,6,6,361.144,361.144,0,1,0,3,3,0,0,0,0,Between -11 and 5
+318,CID 131673872,202.17,84.2,254.0,,14,3,3,0,202.059,202.059,0,3,0,0,0,0,0,0,0,Larger than 6
+319,"Sodium;5-amino-2,3-dihydrophthalazine-1,4-dione;hydride",201.16,84.2,254.0,,14,3,4,0,201.051,201.051,0,3,0,0,0,0,0,0,0,Larger than 6
+320,Zilucoplan,3562.0,1070.0,6980.0,4.8,251,28,57,142,3560.97,3559.97,0,1,0,16,16,0,0,0,0,Between -11 and 5
+321,Defibrotide,444.4,137.0,773.0,1.8,31,4,7,5,444.12,444.12,0,1,0,0,0,0,0,0,0,Between -11 and 5
+322,"7-[(3s,4r)-4-(3-Chlorophenyl)carbonylpyrrolidin-3-Yl]-3h-Quinazolin-4-One",353.8,70.6,567.0,2.1,25,2,4,3,353.093,353.093,0,1,0,2,2,0,0,0,0,Between -11 and 5
+323,Unii-7kyp9tkt70,879.6,484.0,1210.0,,58,15,24,13,879.205,879.205,0,4,0,8,0,8,0,0,0,Larger than 6
+324,"4-acetamidobenzoic acid;9-[(2R,3R,4R,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1H-purin-6-one;1-(dimethylamino)propan-2-ol",1115.2,399.0,658.0,,79,13,22,14,1114.55,1114.55,0,7,0,7,4,3,0,0,0,Larger than 6
+325,"[(1S,4R,6S)-4-(cyclopropylsulfonylcarbamoyl)-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-3,16-diazatricyclo[14.3.0.04,6]nonadec-7-en-18-yl] 4-fluoro-1,3-dihydroisoindole-2-carboxylate",731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,3,2,1,0,1,Between -11 and 5
+326,example 13 [US20210284598A1],257.279,40.5,287.0,2.7,18,1,4,4,257.123,257.123,0,1,0,0,0,0,0,0,0,Between -11 and 5
+327,Stannous protoporphyrin,679.4,102.0,1010.0,,43,2,8,8,680.145,680.145,0,2,0,0,0,0,0,0,0,Larger than 6
+328,"3,6-Di-O-acetyl-2-deoxy-d-glucopyranose",248.23,110.0,276.0,-1.7,17,2,7,9,248.09,248.09,0,1,0,3,3,0,0,0,0,Between -11 and 5
+329,X6 hydrobromide [PMID: 34584244],502.5,168.0,887.0,2.3,36,1,11,11,502.229,502.229,0,1,1,4,4,0,0,0,0,Between -11 and 5
+330,Enoxaparin,1134.9,652.0,2410.0,-10.8,70,15,38,21,1134.01,1134.01,0,1,0,20,0,20,0,0,0,Smaller than -10
+331,Amantadine,151.25,26.0,144.0,2.4,11,1,1,0,151.136,151.136,0,1,0,0,0,0,0,0,0,Between -11 and 5
+332,Amlodipine,408.9,99.9,647.0,3.0,28,2,7,10,408.145,408.145,0,1,0,1,0,1,0,0,0,Between -11 and 5
+333,Bicalutamide,430.4,116.0,750.0,2.3,29,2,9,5,430.061,430.061,0,1,0,1,0,1,0,0,0,Between -11 and 5
+334,Candesartan cilexetil,610.7,143.0,962.0,7.0,45,1,10,13,610.254,610.254,0,1,0,1,0,1,0,0,0,Larger than 6
+335,Formoterol,344.4,90.8,388.0,1.8,25,4,5,8,344.174,344.174,0,1,0,2,0,2,0,0,0,Between -11 and 5
+336,Hydroxychloroquine,335.9,48.4,331.0,3.6,23,2,4,9,335.176,335.176,0,1,0,1,0,1,0,0,0,Between -11 and 5
+337,Ibudilast,230.31,34.4,288.0,3.0,17,0,2,3,230.142,230.142,0,1,0,0,0,0,0,0,0,Between -11 and 5
+338,Lidocaine,234.34,32.299,228.0,2.3,17,1,2,5,234.173,234.173,0,1,0,0,0,0,0,0,0,Between -11 and 5
+339,Modafinil,273.4,79.4,302.0,1.7,19,1,3,5,273.082,273.082,0,1,0,1,0,1,0,0,0,Between -11 and 5
+340,Omeprazole,345.4,96.3,453.0,2.2,24,1,6,5,345.115,345.115,0,1,0,1,0,1,0,0,0,Between -11 and 5
+341,Pentoxifylline,278.31,75.5,426.0,0.3,20,0,4,5,278.138,278.138,0,1,0,0,0,0,0,0,0,Between -11 and 5
+342,Arginine,174.2,128.0,176.0,-4.2,12,4,4,5,174.112,174.112,0,1,0,1,1,0,0,0,0,Between -11 and 5
+343,"4,4'-Diphenylmethane diisocyanate",250.25,58.9,332.0,5.4,19,0,4,4,250.074,250.074,0,1,0,0,0,0,0,0,0,Larger than 6
+344,Carvacrol,150.22,20.2,120.0,3.1,11,1,1,1,150.104,150.104,0,1,0,0,0,0,0,0,0,Between -11 and 5
+345,Silver,107.868,0.0,0.0,,1,0,0,0,106.905,106.905,0,1,0,0,0,0,0,0,0,Larger than 6
+346,Phorbol 12-myristate 13-acetate,616.8,130.0,1150.0,6.5,44,3,8,17,616.398,616.398,0,1,0,8,8,0,0,0,0,Larger than 6
+347,(-)-Mefloquine,378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,2,0,0,0,0,Between -11 and 5
+348,Atorvastatin,558.6,112.0,822.0,5.0,41,4,6,12,558.253,558.253,0,1,0,2,2,0,0,0,0,Between -11 and 5
+349,Propranolol Hydrochloride,295.8,41.5,257.0,,20,3,3,6,295.134,295.134,0,2,0,1,0,1,0,0,0,Larger than 6
+350,Oseltamivir,312.4,90.6,418.0,1.1,22,2,5,8,312.205,312.205,0,1,0,3,3,0,0,0,0,Between -11 and 5
+351,"2,4,6-Trinitro-m-xylene",241.16,138.0,317.0,2.0,17,0,6,0,241.033,241.033,0,1,0,0,0,0,0,0,0,Between -11 and 5
+352,Argatroban,508.6,189.0,887.0,1.3,35,5,8,9,508.247,508.247,0,1,0,4,3,1,0,0,0,Between -11 and 5
+353,Lopinavir,628.8,120.0,940.0,5.9,46,4,5,15,628.362,628.362,0,1,0,4,4,0,0,0,0,Larger than 6
+354,Allopregnanolone,318.5,37.3,500.0,4.9,23,1,2,1,318.256,318.256,0,1,0,8,8,0,0,0,0,Between -11 and 5
+355,Fingolimod,307.5,66.5,258.0,4.2,22,3,3,12,307.251,307.251,0,1,0,0,0,0,0,0,0,Between -11 and 5
+356,Imatinib Mesylate,589.7,149.0,799.0,,42,3,10,7,589.247,589.247,0,2,0,0,0,0,0,0,0,Larger than 6
+357,Meldonium,146.19,52.2,112.0,-2.1,10,1,3,3,146.106,146.106,0,1,0,0,0,0,0,0,0,Between -11 and 5
+358,Ramatroban,416.5,96.8,689.0,2.9,29,2,6,6,416.121,416.121,0,1,0,1,1,0,0,0,0,Between -11 and 5
+359,Ivabradine,468.6,60.5,663.0,2.4,34,0,6,10,468.262,468.262,0,1,0,1,1,0,0,0,0,Between -11 and 5
+360,Moxifloxacin,401.4,82.1,727.0,0.6,29,2,8,4,401.175,401.175,0,1,0,2,2,0,0,0,0,Between -11 and 5
+361,Varespladib,380.4,112.0,589.0,2.8,28,2,5,8,380.137,380.137,0,1,0,0,0,0,0,0,0,Between -11 and 5
+362,Naproxen,230.26,46.5,277.0,3.3,17,1,3,3,230.094,230.094,0,1,0,1,1,0,0,0,0,Between -11 and 5
+363,Dabigatran,471.5,150.0,757.0,1.7,35,4,7,9,471.202,471.202,0,1,0,0,0,0,0,0,0,Between -11 and 5
+364,Senicapoc,323.3,43.1,397.0,4.1,24,1,3,4,323.112,323.112,0,1,0,0,0,0,0,0,0,Between -11 and 5
+365,Povidone iodine,364.95,20.3,120.0,,10,0,1,1,364.877,364.877,0,2,0,0,0,0,0,0,0,Larger than 6
+366,beta-L-Arabinose,150.13,90.2,117.0,-2.5,10,4,5,0,150.053,150.053,0,1,0,4,4,0,0,0,0,Between -11 and 5
+367,Quinidine,324.4,45.6,457.0,2.9,24,1,4,4,324.184,324.184,0,1,0,4,4,0,0,0,0,Between -11 and 5
+368,Pectin,194.14,127.0,205.0,-2.3,13,5,7,1,194.043,194.043,0,1,0,5,5,0,0,0,0,Between -11 and 5
+369,Fluticasone Propionate,500.6,106.0,984.0,4.0,34,1,9,6,500.184,500.184,0,1,0,9,9,0,0,0,0,Between -11 and 5
+370,Decitabine,228.21,121.0,356.0,-1.2,16,3,4,2,228.086,228.086,0,1,0,3,3,0,0,0,0,Between -11 and 5
+371,Bucillamine,223.3,68.4,218.0,0.4,13,4,5,4,223.034,223.034,0,1,0,1,1,0,0,0,0,Between -11 and 5
+372,Canrenoic acid,358.5,74.6,707.0,1.9,26,2,4,3,358.214,358.214,0,1,0,6,6,0,0,0,0,Between -11 and 5
+373,"(S)-[2,8-bis(trifluoromethyl)quinolin-4-yl]-[(2S)-piperidin-2-yl]methanol",378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,2,0,0,0,0,Between -11 and 5
+374,"(-)-(S)-9-Fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyrido(1,2,3-de)-1,4-benzoxazine-6-carboxylic acid, hemihydrate",740.7,148.0,634.0,,53,3,17,4,740.298,740.298,0,3,0,2,2,0,0,0,0,Larger than 6
+375,"(S,S)-Formoterol",344.4,90.8,388.0,1.8,25,4,5,8,344.174,344.174,0,1,0,2,2,0,0,0,0,Between -11 and 5
+376,Glatiramer acetate,623.7,374.0,519.0,,43,12,18,13,623.301,623.301,0,5,0,4,4,0,0,0,0,Larger than 6
+377,Isoquercetin,464.4,207.0,758.0,0.4,33,8,12,4,464.095,464.095,0,1,0,5,5,0,0,0,0,Between -11 and 5
+378,Pitavastatin,421.5,90.6,631.0,3.5,31,3,6,8,421.169,421.169,0,1,0,2,2,0,1,1,0,Between -11 and 5
+379,Deoxy-methyl-arginine,172.23,108.0,174.0,-1.8,12,3,3,5,172.132,172.132,0,1,0,1,1,0,0,0,0,Between -11 and 5
+380,Dexmedetomidine,200.28,28.7,205.0,3.1,15,1,1,2,200.131,200.131,0,1,0,1,1,0,0,0,0,Between -11 and 5
+381,Nafamostat mesylate,539.6,266.0,645.0,,36,6,10,5,539.114,539.114,0,3,0,0,0,0,0,0,0,Larger than 6
+382,Bromhexine Hydrochloride,412.59,29.3,256.0,,19,2,2,3,411.974,409.976,0,2,0,0,0,0,0,0,0,Larger than 6
+383,Tenofovir Disoproxil Fumarate,635.5,260.0,817.0,,43,3,18,19,635.184,635.184,0,2,0,1,1,0,1,1,0,Larger than 6
+384,Eritoran,1313.7,294.0,1900.0,15.4,89,7,19,59,1312.84,1312.84,0,1,0,11,11,0,1,1,0,Larger than 6
+385,Icatibant acetate,1364.6,589.0,2750.0,,96,16,20,30,1363.68,1363.68,0,2,0,12,12,0,0,0,0,Larger than 6
+386,Dalcetrapib,389.6,71.5,481.0,7.1,27,1,3,9,389.239,389.239,0,1,0,0,0,0,0,0,0,Larger than 6
+387,"7-[[(2Z)-2-ethoxyimino-2-[5-(phosphonoamino)-1,2,4-thiadiazol-3-yl]acetyl]amino]-3-[[4-(1-methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid",685.7,328.0,1220.0,1.6,43,5,17,11,685.018,685.018,1,1,0,2,0,2,1,1,0,Between -11 and 5
+388,Icosapent ethyl,330.5,26.3,425.0,6.3,24,0,2,15,330.256,330.256,0,1,0,0,0,0,5,5,0,Larger than 6
+389,"1-Piperazinecarboxamide, 4-methyl-N-((1S)-2-oxo-2-(((1S)-1-(2-phenylethyl)-3-(phenylsulfonyl)-2-propenyl)amino)-1-(phenylmethyl)ethyl)-",574.7,107.0,939.0,4.1,41,2,5,11,574.261,574.261,0,1,0,2,2,0,1,1,0,Between -11 and 5
+390,Remimazolam,439.3,69.4,601.0,3.4,28,0,5,5,438.069,438.069,0,1,0,1,1,0,0,0,0,Between -11 and 5
+391,Azithromycin Monohydrate,767.0,181.0,1150.0,,53,6,15,7,766.519,766.519,0,2,0,18,18,0,0,0,0,Larger than 6
+392,Danoprevir,731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,5,0,1,1,0,Between -11 and 5
+393,Mitoquinone,583.7,52.6,886.0,9.4,42,0,4,16,583.298,583.298,1,1,0,0,0,0,0,0,0,Larger than 6
+394,Belnacasan,509.0,140.0,818.0,2.3,35,3,7,8,508.209,508.209,0,1,0,4,4,0,0,0,0,Between -11 and 5
+395,Apilimod mesylate,610.7,210.0,637.0,,41,3,14,8,610.188,610.188,0,3,0,0,0,0,1,1,0,Larger than 6
+396,Losmapimod,383.5,71.1,573.0,3.8,28,2,4,6,383.201,383.201,0,1,0,0,0,0,0,0,0,Between -11 and 5
+397,Emtricitabine and tenofovir disoproxil fumarate,882.8,374.0,1190.0,,59,5,23,21,882.227,882.227,0,3,0,3,3,0,1,1,0,Larger than 6
+398,Dapansutrile,133.17,66.3,190.0,-0.7,8,0,3,2,133.02,133.02,0,1,0,0,0,0,0,0,0,Between -11 and 5
+399,"(S)-[2,8-bis(trifluoromethyl)quinolin-4-yl]-piperidin-2-ylmethanol",378.31,45.2,483.0,3.6,26,2,9,2,378.117,378.117,0,1,0,2,1,1,0,0,0,Between -11 and 5
+400,Tridecactide,1623.8,659.0,3240.0,-6.2,115,23,24,50,1622.77,1622.77,0,1,0,12,12,0,0,0,0,Between -11 and 5
+401,Aviptadil,3326.8,1470.0,7580.0,-15.9,234,51,51,115,3325.74,3324.74,0,1,0,31,31,0,0,0,0,Smaller than -10
+402,Pamapimod,406.4,108.0,591.0,2.4,29,3,9,8,406.145,406.145,0,1,0,0,0,0,0,0,0,Between -11 and 5
+403,Sodium pyruvate,110.04,57.2,88.2,,7,0,3,1,109.998,109.998,0,2,0,0,0,0,0,0,0,Larger than 6
+404,Brequinar sodium,397.3,53.0,557.0,,29,0,5,3,397.089,397.089,0,2,0,0,0,0,0,0,0,Larger than 6
+405,Montelukast Sodium,608.2,98.6,898.0,,42,1,5,12,607.192,607.192,0,2,0,1,1,0,1,1,0,Larger than 6
+406,Sivelestat sodium anhydrous,456.4,150.0,738.0,,31,2,8,9,456.097,456.097,0,2,0,0,0,0,0,0,0,Larger than 6
+407,Methylprednisolone sodium succinate,496.5,141.0,988.0,,35,2,8,7,496.207,496.207,0,2,0,8,8,0,0,0,0,Larger than 6
+408,Bromelains,248.25,81.6,344.0,,17,1,3,4,248.114,248.114,0,2,0,2,0,2,0,0,0,Larger than 6
+409,Fostamatinib disodium,732.5,198.0,893.0,,48,8,21,9,732.176,732.176,0,9,0,0,0,0,0,0,0,Larger than 6
+410,Phosphate-Buffered Saline,411.04,164.0,96.3,,17,3,10,0,409.765,409.765,0,9,0,0,0,0,0,0,0,Larger than 6
+411,4'-C-Azido-2'-deoxy-2'-fluoro-b-D-arabinocytidine,286.22,123.0,533.0,-0.8,20,3,7,3,286.083,286.083,0,1,0,4,0,4,1,0,1,Between -11 and 5
+412,5-Chloro-2-([(2-([3-(furan-2-yl)phenyl]amino)-2-oxoethoxy)acetyl]amino)benzoic acid,428.8,118.0,618.0,3.3,30,3,6,8,428.078,428.078,0,1,0,0,0,0,0,0,0,Between -11 and 5
+413,"[(2R,3S,4R)-4-acetyloxy-3,6-dihydroxyoxan-2-yl]methyl acetate",248.23,102.0,290.0,-1.0,17,2,7,5,248.09,248.09,0,1,0,4,3,1,0,0,0,Between -11 and 5
+414,Ketone Ester,176.21,66.8,135.0,-0.1,12,2,4,6,176.105,176.105,0,1,0,2,2,0,0,0,0,Between -11 and 5
+415,CID 45114162,520.6,152.0,761.0,,34,2,10,9,520.123,520.123,1,2,0,2,2,0,1,1,0,Larger than 6
+416,Alvelestat,545.5,123.0,1100.0,2.5,38,1,9,6,545.134,545.134,0,1,0,0,0,0,0,0,0,Between -11 and 5
+417,Emvododstat,467.3,54.6,651.0,6.3,32,1,3,4,466.085,466.085,0,1,0,1,1,0,0,0,0,Larger than 6
+418,8-chloro-N-[4-(trifluoromethoxy)phenyl]quinolin-2-amine,338.71,34.2,388.0,5.9,23,1,6,3,338.043,338.043,0,1,0,0,0,0,0,0,0,Larger than 6
+419,Riamilovir sodium dihydrate,286.2,152.0,262.0,,18,2,10,1,286.01,286.01,0,4,0,0,0,0,0,0,0,Larger than 6
+420,Doxycycline hyclate,545.0,203.0,958.0,,37,9,11,2,544.182,544.182,0,4,0,6,6,0,0,0,0,Larger than 6
+421,Budesonide and formoterol fumarate dihydrate,774.9,184.0,1250.0,,56,6,11,12,774.409,774.409,0,2,0,11,10,1,0,0,0,Larger than 6
+422,Vafidemstat,336.4,86.2,410.0,2.2,25,2,6,7,336.159,336.159,0,1,0,2,2,0,0,0,0,Between -11 and 5
+423,Ledipasvir,889.0,175.0,1820.0,7.4,65,4,10,12,888.413,888.413,0,1,0,6,6,0,0,0,0,Larger than 6
+424,Telacebec,557.0,58.9,796.0,7.9,39,1,7,7,556.185,556.185,0,1,0,0,0,0,0,0,0,Larger than 6
+425,Acebilustat,481.5,79.0,728.0,2.2,36,1,7,8,481.2,481.2,0,1,0,2,2,0,0,0,0,Between -11 and 5
+426,Quinidine monohydrate,342.4,46.6,457.0,,25,2,5,4,342.194,342.194,0,2,0,4,4,0,0,0,0,Larger than 6
+427,Dexamethasone phosphate disodium,518.4,141.0,973.0,,34,4,9,4,518.146,518.146,0,3,0,8,8,0,0,0,0,Larger than 6
+428,Montmorillonite,360.31,141.0,18.3,,18,1,12,0,359.825,359.825,0,10,0,0,0,0,0,0,0,Larger than 6
+429,methyl N-[(2R)-2-[2-[5-[4-[4-[2-[1-[(2S)-2-(methoxycarbonylamino)-3-methyl-butanoyl]pyrrolidin-2-yl]-1H-imidazol-5-yl]phenyl]phenyl]-1H-imidazol-2-yl]pyrrolidine-1-carbonyl]-3-methyl-butyl]carbamate,752.9,175.0,1300.0,5.0,55,4,8,14,752.401,752.401,0,1,0,4,2,2,0,0,0,Between -11 and 5
+430,Nadide sodium,685.4,324.0,1110.0,,45,6,18,11,685.091,685.091,0,2,0,8,8,0,0,0,0,Larger than 6
+431,"(6R,7R)-7-[[(2E)-2-ethoxyimino-2-[5-(phosphonoamino)-1,2,4-thiadiazol-3-yl]acetyl]amino]-3-[[4-(1-methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate",684.7,330.0,1210.0,2.3,43,4,17,10,684.01,684.01,0,1,0,2,2,0,1,1,0,Between -11 and 5
+432,"3-Deoxy-3-[4-(3-Fluorophenyl)-1h-1,2,3-Triazol-1-Yl]-Beta-D-Galactopyranosyl 3-Deoxy-3-[4-(3-Fluorophenyl)-1h-1,2,3-Triazol-1-Yl]-1-Thio-Beta-D-Galactopyranoside",648.6,227.0,903.0,0.2,45,6,15,8,648.181,648.181,0,1,0,10,10,0,0,0,0,Between -11 and 5
+433,"N-[(2S,3S,4R)-1-(alpha-D-galactopyranosyloxy)-3,4-dihydroxyoctadecan-2-yl]undecanamide",647.9,169.0,700.0,7.9,45,7,9,29,647.497,647.497,0,1,0,8,8,0,0,0,0,Larger than 6
+434,Brensocatib,420.5,104.0,699.0,2.0,31,2,6,5,420.18,420.18,0,1,0,2,2,0,0,0,0,Between -11 and 5
+435,Ezurpimtrostat,423.0,40.2,520.0,5.9,30,2,4,6,422.224,422.224,0,1,0,0,0,0,0,0,0,Larger than 6
+436,Dexamethasone 21-phosphate disodium salt,548.5,141.0,973.0,,36,4,9,4,548.193,548.193,0,4,0,8,8,0,0,0,0,Larger than 6
+437,Enpatoran,320.31,65.9,472.0,2.7,23,1,7,1,320.125,320.125,0,1,0,2,2,0,0,0,0,Between -11 and 5
+438,H-D-Lys-D-Leu-D-Leu-D-Leu-D-Leu-D-Lys-D-Leu-D-Leu-D-Leu-D-Leu-D-Lys-D-Leu-D-Leu-D-Leu-D-Leu-D-Lys-D-Leu-D-Leu-D-Leu-D-Leu-D-Lys-OH,2469.4,775.0,4880.0,11.9,174,27,28,93,2468.83,2467.83,0,1,0,21,21,0,0,0,0,Larger than 6
+439,CID 131844884,1738.2,873.0,3450.0,,101,19,52,30,1736.85,1736.85,0,11,0,25,25,0,0,0,0,Larger than 6
+440,Folic Acid,441.4,209.0,767.0,-1.1,32,6,10,9,441.14,441.14,0,1,0,1,1,0,0,0,0,Between -11 and 5
+441,Inosine pranobex,1115.2,399.0,658.0,,79,13,22,14,1114.55,1114.55,0,7,0,7,4,3,0,0,0,Larger than 6
+442,Eltrombopag,442.5,115.0,812.0,5.4,33,3,7,5,442.164,442.164,0,1,0,0,0,0,0,0,0,Larger than 6
+443,Rintatolimod,995.6,517.0,1600.0,,65,15,27,12,995.135,995.135,0,3,0,12,12,0,0,0,0,Larger than 6
+444,Normosang,792.7,260.0,1180.0,,55,8,10,13,792.305,792.305,2,3,0,1,1,0,0,0,0,Larger than 6
+445,Artecom,1294.4,485.0,1380.0,,84,15,31,12,1293.31,1293.31,0,6,0,8,8,0,0,0,0,Larger than 6
+446,Imunovir; Delimmun; Groprinosin;Inosine pranobex,1115.2,399.0,658.0,,79,13,22,14,1114.55,1114.55,0,7,0,7,0,7,0,0,0,Larger than 6
+447,"1-(4-fluorobenzene-6-id-1-yl)-N-phenylmethanimine;iridium(3+);1,2,3,4,5-pentamethylcyclopenta-1,3-diene;chloride",561.1,12.4,601.0,,27,0,5,1,561.121,561.121,0,4,0,0,0,0,0,0,0,Larger than 6
+448,EIDD-2801,329.31,141.0,534.0,-0.8,23,4,7,6,329.122,329.122,0,1,0,4,4,0,0,0,0,Between -11 and 5
+449,CID 154701548,287.21,143.0,435.0,,18,3,8,1,287.017,287.017,0,4,0,0,0,0,0,0,0,Larger than 6
+450,"alpha-D-Glucopyranoside, methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-alpha-D-glucopyranosyl-(1-->4)-O-beta-D-glucopyranuronosyl-(1-->4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-alpha-D-glucopyranosyl-(1-->4)-O-2-O-sulfo-alpha-L-idopyranuronosyl-(1-->4)-2-deoxy-2-(sulfoamino)-, 6-(hydrogen sulfate), sodium salt (1:10)",1531.3,873.0,3450.0,,92,19,52,30,1529.94,1529.94,0,2,0,25,25,0,0,0,0,Larger than 6
+451,"propan-2-yl (2S)-2-[[[(2R,3R,4R,5R)-5-[2-amino-6-(methylamino)purin-9-yl]-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate;sulfuric acid",679.6,268.0,1000.0,,45,6,18,12,679.184,679.184,0,2,0,6,5,1,0,0,0,Larger than 6
+452,Ensitrelvir,531.9,114.0,919.0,2.5,37,1,8,6,531.115,531.115,0,1,0,0,0,0,0,0,0,Between -11 and 5
+453,"3,4-Methylenedioxymethamphetamine",193.24,30.5,186.0,2.2,14,1,3,3,193.11,193.11,0,1,0,1,0,1,0,0,0,Between -11 and 5
+454,Acetaminophen,151.16,49.3,139.0,0.5,11,2,2,1,151.063,151.063,0,1,0,0,0,0,0,0,0,Between -11 and 5
+455,Amiodarone,645.3,42.7,547.0,7.6,31,0,4,11,645.024,645.024,0,1,0,0,0,0,0,0,0,Larger than 6
+456,Verapamil,454.6,64.0,606.0,3.8,33,0,6,13,454.283,454.283,0,1,0,1,0,1,0,0,0,Between -11 and 5
+457,Candesartan,440.5,119.0,660.0,4.1,33,2,7,7,440.16,440.16,0,1,0,0,0,0,0,0,0,Between -11 and 5
+458,Chlordiazepoxide,299.75,48.2,580.0,2.4,21,1,3,1,299.083,299.083,0,1,0,0,0,0,0,0,0,Between -11 and 5
+459,Chloroquine,319.9,28.2,309.0,4.6,22,1,3,8,319.182,319.182,0,1,0,1,0,1,0,0,0,Between -11 and 5
+460,Deferoxamine,560.7,206.0,739.0,-2.1,39,6,9,23,560.353,560.353,0,1,0,0,0,0,0,0,0,Between -11 and 5
+461,Famotidine,337.5,238.0,469.0,-0.6,20,4,8,7,337.045,337.045,0,1,0,0,0,0,1,0,1,Between -11 and 5
+462,Fenofibrate,360.8,52.6,458.0,5.2,25,0,4,7,360.113,360.113,0,1,0,0,0,0,0,0,0,Larger than 6
+463,Ketamine,237.72,29.1,269.0,2.2,16,1,2,2,237.092,237.092,0,1,0,1,0,1,0,0,0,Between -11 and 5
+464,Lansoprazole,369.4,87.1,480.0,2.8,25,1,8,5,369.076,369.076,0,1,0,1,0,1,0,0,0,Between -11 and 5
+465,Metformin,129.16,91.5,132.0,-1.3,9,3,1,2,129.101,129.101,0,1,0,0,0,0,0,0,0,Between -11 and 5
+466,Nafamostat,347.4,141.0,552.0,2.0,26,4,4,5,347.138,347.138,0,1,0,0,0,0,0,0,0,Between -11 and 5
+467,Palmitoylethanolamide,299.5,49.3,219.0,6.2,21,2,2,16,299.282,299.282,0,1,0,0,0,0,0,0,0,Larger than 6
+468,Pioglitazone,356.4,93.6,466.0,3.8,25,1,5,7,356.119,356.119,0,1,0,1,0,1,0,0,0,Between -11 and 5
+469,Propofol,178.27,20.2,135.0,3.8,13,1,1,2,178.136,178.136,0,1,0,0,0,0,0,0,0,Between -11 and 5
+470,Sevoflurane,200.05,9.2,121.0,2.8,12,0,8,2,200.007,200.007,0,1,0,0,0,0,0,0,0,Between -11 and 5
+471,Tranexamic acid,157.21,63.3,139.0,-2.0,11,2,3,2,157.11,157.11,0,1,0,0,0,0,0,0,0,Between -11 and 5
+472,Prednisolone,360.4,94.8,724.0,1.6,26,3,5,2,360.194,360.194,0,1,0,7,7,0,0,0,0,Between -11 and 5
+473,Levothyroxine,776.87,92.8,420.0,2.4,24,3,5,5,776.687,776.687,0,1,0,1,1,0,0,0,0,Between -11 and 5
+474,Mannitol,182.17,121.0,105.0,-3.1,12,6,6,5,182.079,182.079,0,1,0,4,4,0,0,0,0,Between -11 and 5
+475,Dexamethasone phosphate,472.4,141.0,973.0,0.1,32,4,9,4,472.166,472.166,0,1,0,8,8,0,0,0,0,Between -11 and 5
+476,Citrulline,175.19,118.0,171.0,-4.3,12,4,4,5,175.096,175.096,0,1,0,1,1,0,0,0,0,Between -11 and 5
+477,Hesperidin,610.6,234.0,940.0,-1.1,43,8,15,7,610.19,610.19,0,1,0,11,11,0,0,0,0,Between -11 and 5
+478,Zinc Citrate,574.3,281.0,211.0,,29,2,14,4,571.791,569.794,0,5,0,0,0,0,0,0,0,Larger than 6
+479,Zinc Acetate,183.5,80.3,25.5,,9,0,4,0,181.956,181.956,0,3,0,0,0,0,0,0,0,Larger than 6
+480,Canrenone,340.5,43.4,719.0,2.7,25,0,3,0,340.204,340.204,0,1,0,6,6,0,0,0,0,Between -11 and 5
+481,Hydrocortisone hemisuccinate,462.5,138.0,908.0,1.8,33,3,8,7,462.225,462.225,0,1,0,7,7,0,0,0,0,Between -11 and 5
+482,Methylprednisolone hemisuccinate,474.5,138.0,981.0,2.2,34,3,8,7,474.225,474.225,0,1,0,8,8,0,0,0,0,Between -11 and 5
+483,Azo rubin S,458.5,170.0,880.0,3.3,31,3,9,4,458.024,458.024,0,1,0,0,0,0,0,0,0,Between -11 and 5
+484,"Zinc, bis(D-gluconato-kappaO1,kappaO2)-, (T-4)-",457.7,277.0,385.0,,27,12,14,10,456.046,456.046,0,3,0,8,8,0,0,0,0,Larger than 6
+485,2-acetamido-2-deoxy-beta-D-glucopyranose,221.21,119.0,235.0,-1.7,15,5,6,2,221.09,221.09,0,1,0,5,5,0,0,0,0,Between -11 and 5
+486,Zinc Sulfate,161.4,88.6,62.2,,6,0,4,0,159.881,159.881,0,2,0,0,0,0,0,0,0,Larger than 6
+487,Sodium Thiosulfate,158.11,104.0,82.6,,7,0,4,0,157.908,157.908,0,3,0,0,0,0,0,0,0,Larger than 6
+488,Diacerein,368.3,124.0,683.0,1.9,27,1,8,5,368.053,368.053,0,1,0,0,0,0,0,0,0,Between -11 and 5
+489,Levamisole,204.29,40.9,246.0,1.8,14,0,2,1,204.072,204.072,0,1,0,1,1,0,0,0,0,Between -11 and 5
+490,Cromolyn sodium,512.299,172.0,824.0,,36,1,11,6,512.033,512.033,0,3,0,0,0,0,0,0,0,Larger than 6
+491,Calcium dobesilate,418.4,212.0,228.0,,25,4,10,0,417.934,417.934,0,3,0,0,0,0,0,0,0,Larger than 6
+492,Amoxicillin,365.4,158.0,590.0,-2.0,25,4,7,4,365.105,365.105,0,1,0,4,4,0,0,0,0,Between -11 and 5
+493,Tramadol,263.37,32.7,282.0,2.6,19,1,3,4,263.189,263.189,0,1,0,2,2,0,0,0,0,Between -11 and 5
+494,Almitrine,477.6,69.2,602.0,5.6,35,2,9,10,477.245,477.245,0,1,0,0,0,0,0,0,0,Larger than 6
+495,Pirfenidone,185.22,20.3,285.0,1.9,14,0,1,1,185.084,185.084,0,1,0,0,0,0,0,0,0,Between -11 and 5
+496,Piperacillin,517.6,182.0,982.0,0.5,36,3,8,6,517.163,517.163,0,1,0,4,4,0,0,0,0,Between -11 and 5
+497,Captopril,217.29,58.6,244.0,0.3,14,2,4,3,217.077,217.077,0,1,0,2,2,0,0,0,0,Between -11 and 5
+498,Nicorandil,211.17,97.0,228.0,0.8,15,1,5,4,211.059,211.059,0,1,0,0,0,0,0,0,0,Between -11 and 5
+499,Brequinar,375.4,50.2,551.0,5.6,28,1,5,3,375.107,375.107,0,1,0,0,0,0,0,0,0,Larger than 6
+500,Amantadine hydrochloride,187.71,26.0,144.0,,12,2,1,0,187.113,187.113,0,2,0,0,0,0,0,0,0,Larger than 6
+501,Chloroquine Phosphate,515.9,184.0,359.0,,32,7,11,8,515.135,515.135,0,3,0,1,0,1,0,0,0,Larger than 6
+502,Epigallocatechin Gallate,458.4,197.0,667.0,1.2,33,8,11,4,458.085,458.085,0,1,0,2,2,0,0,0,0,Between -11 and 5
+503,Artemisinin,282.33,54.0,452.0,2.8,20,0,5,0,282.147,282.147,0,1,0,7,7,0,0,0,0,Between -11 and 5
+504,"1-((2S,3R,4S,5S)-3-Fluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione",260.22,99.1,413.0,-0.9,18,3,6,2,260.081,260.081,0,1,0,4,4,0,0,0,0,Between -11 and 5
+505,Bis (2-Carboxyethylgermanium)sesquioxide,339.4,118.0,259.0,,15,2,7,8,339.886,341.885,0,1,0,0,0,0,0,0,0,Larger than 6
+506,Clarithromycin,748.0,183.0,1190.0,3.2,52,4,14,8,747.477,747.477,0,1,0,18,18,0,0,0,0,Between -11 and 5
+507,"2,6-Difluorophenol",130.09,20.2,87.1,2.0,9,1,3,0,130.023,130.023,0,1,0,0,0,0,0,0,0,Between -11 and 5
+508,Moxifloxacin Hydrochloride,437.9,82.1,727.0,,30,3,8,4,437.152,437.152,0,2,0,2,2,0,0,0,0,Larger than 6
+509,Levofloxacin,361.4,73.3,634.0,-0.4,26,1,8,2,361.144,361.144,0,1,0,1,1,0,0,0,0,Between -11 and 5
+510,Olmesartan,446.5,130.0,656.0,3.2,33,3,7,8,446.207,446.207,0,1,0,0,0,0,0,0,0,Between -11 and 5
+511,Tetrasul sulfoxide,340.0,36.3,307.0,5.2,18,0,2,2,339.886,337.889,0,1,0,1,0,1,0,0,0,Larger than 6
+512,Ramelteon,259.339,38.3,331.0,2.7,19,1,2,4,259.157,259.157,0,1,0,1,1,0,0,0,0,Between -11 and 5
+513,Darunavir,547.7,149.0,853.0,2.9,38,3,9,12,547.235,547.235,0,1,0,5,5,0,0,0,0,Between -11 and 5
+514,Galactomannan,504.4,269.0,641.0,-6.3,34,11,16,7,504.169,504.169,0,1,0,15,15,0,0,0,0,Between -11 and 5
+515,Mometasone furoate,521.4,93.8,1020.0,3.9,35,1,6,5,520.142,520.142,0,1,0,8,8,0,0,0,0,Between -11 and 5
+516,Rosuvastatin,481.5,149.0,767.0,1.6,33,3,10,10,481.168,481.168,0,1,0,2,2,0,1,1,0,Between -11 and 5
+517,Tenofovir,287.21,136.0,354.0,-1.6,19,3,8,5,287.078,287.078,0,1,0,1,1,0,0,0,0,Between -11 and 5
+518,3-Cyclopentyl-1-(piperazin-1-yl)propan-1-one,210.32,32.299,206.0,1.6,15,1,2,3,210.173,210.173,0,1,0,0,0,0,0,0,0,Between -11 and 5
+519,Azithromycin Dihydrate,785.0,182.0,1150.0,,54,7,16,7,784.53,784.53,0,3,0,18,18,0,0,0,0,Larger than 6
+520,"D-Alanine, N-methylglycyl-L-arginyl-L-valyl-L-tyrosyl-L-isoleucyl-L-histidyl-L-prolyl-",926.1,358.0,1690.0,-2.3,66,12,13,26,925.513,925.513,0,1,0,8,8,0,0,0,0,Between -11 and 5
+521,Arformoterol,344.4,90.8,388.0,1.8,25,4,5,8,344.174,344.174,0,1,0,2,2,0,0,0,0,Between -11 and 5
+522,3-Hydroxybutyrate,103.1,60.4,63.8,0.1,7,1,3,1,103.04,103.04,-1,1,0,1,0,1,0,0,0,Between -11 and 5
+523,Sitagliptin,407.31,77.0,566.0,0.7,28,1,10,4,407.118,407.118,0,1,0,1,1,0,0,0,0,Between -11 and 5
+524,Best,572.3,101.0,412.0,,18,0,4,0,573.618,575.617,0,1,0,0,0,0,0,0,0,Larger than 6
+525,Eprosartan,424.5,121.0,618.0,4.5,30,2,6,10,424.146,424.146,0,1,0,0,0,0,1,1,0,Between -11 and 5
+526,Isotretinoin,300.4,37.3,567.0,6.3,22,1,2,5,300.209,300.209,0,1,0,0,0,0,4,4,0,Larger than 6
+527,Epoprostenol,352.5,87.0,485.0,2.9,25,3,5,10,352.225,352.225,0,1,0,5,5,0,2,2,0,Between -11 and 5
+528,Camostat mesylate,494.5,200.0,695.0,,34,3,9,9,494.147,494.147,0,2,0,0,0,0,0,0,0,Larger than 6
+529,Fluticasone,444.5,99.9,861.0,3.2,30,2,8,3,444.158,444.158,0,1,0,9,9,0,0,0,0,Between -11 and 5
+530,Tenofovir disoproxil,519.4,185.0,698.0,1.6,35,1,14,17,519.173,519.173,0,1,0,1,1,0,0,0,0,Between -11 and 5
+531,Refanalin,176.24,56.9,170.0,2.2,12,1,2,2,176.041,176.041,0,1,0,0,0,0,1,1,0,Between -11 and 5
+532,Sulodexide,295.29,115.0,363.0,0.2,21,3,8,4,295.128,295.128,0,1,0,4,4,0,0,0,0,Between -11 and 5
+533,Metampicillin,361.4,124.0,603.0,3.0,25,2,6,5,361.11,361.11,0,1,0,4,4,0,0,0,0,Between -11 and 5
+534,Ciclesonide,540.7,99.1,1100.0,5.3,39,1,7,6,540.309,540.309,0,1,0,9,9,0,0,0,0,Larger than 6
+535,Dutasteride,528.5,58.2,964.0,5.4,37,2,8,2,528.221,528.221,0,1,0,7,7,0,0,0,0,Larger than 6
+536,Prasugrel,373.4,74.8,555.0,3.6,26,0,6,6,373.115,373.115,0,1,0,1,0,1,0,0,0,Between -11 and 5
+537,Almitrine mesylate,669.8,195.0,694.0,,45,4,15,10,669.221,669.221,0,3,0,0,0,0,0,0,0,Larger than 6
+538,Ile-Ser,218.25,113.0,232.0,-3.3,15,4,5,6,218.127,218.127,0,1,0,3,3,0,0,0,0,Between -11 and 5
+539,butyl (3-(4-((1H-imidazol-1-yl)methyl)phenyl)-5-isobutylthiophen-2-yl)sulfonylcarbamate,475.6,127.0,690.0,5.3,32,1,6,11,475.16,475.16,0,1,0,0,0,0,0,0,0,Larger than 6
+540,Vidofludimus,355.4,75.6,576.0,3.4,26,2,5,5,355.122,355.122,0,1,0,0,0,0,0,0,0,Between -11 and 5
+541,Reparixin,283.39,71.6,389.0,2.9,19,1,3,5,283.124,283.124,0,1,0,1,1,0,0,0,0,Between -11 and 5
+542,Ticagrelor,522.6,164.0,736.0,2.0,36,4,12,10,522.186,522.186,0,1,0,6,6,0,0,0,0,Between -11 and 5
+543,Anhydrous Ceftriaxone Sodium,577.6,288.0,1110.0,,37,4,13,8,577.036,577.036,0,2,0,2,2,0,1,1,0,Larger than 6
+544,Tradipitant,587.9,73.6,865.0,6.2,41,0,11,6,587.095,587.095,0,1,0,0,0,0,0,0,0,Larger than 6
+545,CID 9939931,701.6,316.0,819.0,,47,12,9,18,700.261,700.261,0,2,0,4,4,0,2,2,0,Larger than 6
+546,1-Palmitoyl-2-linoleoyl-3-acetyl-rac-glycerol,635.0,78.9,744.0,14.1,45,0,6,36,634.517,634.517,0,1,0,1,0,1,2,2,0,Larger than 6
+547,Zinforo,744.7,368.0,1240.0,,47,5,19,10,744.031,744.031,0,2,0,2,2,0,1,1,0,Larger than 6
+548,Linagliptin,472.5,114.0,885.0,1.9,35,1,7,4,472.234,472.234,0,1,0,1,1,0,0,0,0,Between -11 and 5
+549,"2-(2-Chloro-4-iodophenylamino)-3,4-difluorobenzoic acid",409.55,49.3,363.0,5.0,20,2,5,3,408.918,408.918,0,1,0,0,0,0,0,0,0,Between -11 and 5
+550,Edoxaban,548.1,165.0,880.0,1.4,37,3,8,5,547.177,547.177,0,1,0,3,3,0,0,0,0,Between -11 and 5
+551,Transcrocetinate sodium,372.4,80.3,597.0,,26,0,4,6,372.131,372.131,0,3,0,0,0,0,7,7,0,Larger than 6
+552,Galidesivir,265.27,140.0,334.0,-2.1,19,6,7,2,265.117,265.117,0,1,0,4,4,0,0,0,0,Between -11 and 5
+553,"4,5-Dihydro-3-phenyl-5-isoxazoleacetic acid",205.21,58.9,269.0,1.3,15,1,4,3,205.074,205.074,0,1,0,1,0,1,0,0,0,Between -11 and 5
+554,5-Cholesten-3beta-25-diol-3-sulfate,482.7,92.2,858.0,6.4,33,2,5,7,482.307,482.307,0,1,0,8,8,0,0,0,0,Larger than 6
+555,"disodium;(6R,7R)-7-[[(2Z)-2-(2-amino-1,3-thiazol-4-yl)-2-methoxyiminoacetyl]amino]-3-[(2-methyl-6-oxido-5-oxo-1,2,4-triazin-3-yl)sulfanylmethyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate;hydrate",616.6,298.0,1120.0,,39,3,15,7,616.021,616.021,0,4,0,2,2,0,1,1,0,Larger than 6
+556,Losartan potassium,461.0,77.7,526.0,,31,1,6,8,460.118,460.118,0,2,0,0,0,0,0,0,0,Larger than 6
+557,Lopinavir and ritonavir,1349.7,322.0,1980.0,,96,8,14,33,1348.68,1348.68,0,2,0,8,8,0,0,0,0,Larger than 6
+558,"N-[5-[[2-(2,6-dimethylphenoxy)acetyl]amino]-4-hydroxy-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxo-1,3-diazinan-1-yl)butanamide;1,3-thiazol-5-ylmethyl N-[(2S,3S,5S)-3-hydroxy-5-[[(2S)-3-methyl-2-[[methyl-[(2-propan-2-yl-1,3-thiazol-4-yl)methyl]carbamoyl]amino]butanoyl]amino]-1,6-diphenylhexan-2-yl]carbamate",1349.7,322.0,1980.0,,96,8,14,33,1348.68,1348.68,0,2,0,8,4,4,0,0,0,Larger than 6
+559,"S-[2-[3-[[(2R)-4-[[[(2R,3S,4R,5R)-5-(6-aminopurin-9-yl)-4-hydroxy-3-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl]oxy-2-hydroxy-3,3-dimethylbutanoyl]amino]propanoylamino]ethyl] (2S)-2-[4-(2-methylpropyl)phenyl]propanethioate",955.8,389.0,1660.0,-1.9,62,9,22,24,955.235,955.235,0,1,0,6,6,0,0,0,0,Between -11 and 5
+560,Bivalirudin,2180.3,902.0,4950.0,-7.1,155,28,35,67,2179.99,2178.99,0,1,0,16,16,0,0,0,0,Between -11 and 5
+561,Thymalfasin,3108.3,1460.0,7190.0,-24.0,217,49,59,111,3107.51,3106.5,0,1,0,32,32,0,0,0,0,Smaller than -10
+562,CID 16219160,397.6,74.6,707.0,,27,2,4,3,397.178,397.178,0,2,0,6,6,0,0,0,0,Larger than 6
+563,Sodium valproate,166.19,40.1,98.3,,11,0,2,5,166.097,166.097,0,2,0,0,0,0,0,0,0,Larger than 6
+564,"N,N-Diethyl-(2-(4-(2-(18F)fluoroethoxy)phenyl)-5,7-dimethylpyrazolo(1,5-A)pyrimidine-3-YL)acetamide",397.5,59.7,525.0,3.2,29,0,5,8,397.214,397.214,0,1,1,0,0,0,0,0,0,Between -11 and 5
+565,Amoxicillin sodium,387.4,161.0,596.0,,26,3,7,4,387.086,387.086,0,2,0,4,4,0,0,0,0,Larger than 6
+566,Potassium canrenoate,396.6,77.4,713.0,,27,1,4,3,396.17,396.17,0,2,0,6,6,0,0,0,0,Larger than 6
+567,Hydrocortisone sodium succinate,484.5,141.0,915.0,,34,2,8,7,484.207,484.207,0,2,0,7,7,0,0,0,0,Larger than 6
+568,Piperacillin-tazobactam,839.8,315.0,1560.0,,57,3,15,9,839.198,839.198,0,3,0,7,7,0,0,0,0,Larger than 6
+569,Antroquinonol,390.6,55.8,648.0,5.8,28,1,4,10,390.277,390.277,0,1,0,3,3,0,2,2,0,Larger than 6
+570,Brilacidin,936.9,314.0,1560.0,0.3,66,10,18,20,936.394,936.394,0,1,0,2,2,0,0,0,0,Between -11 and 5
+571,Ruxolitinib phosphate,404.4,161.0,503.0,,28,4,8,4,404.136,404.136,0,2,0,1,1,0,0,0,0,Larger than 6
+572,Baricitinib,371.4,129.0,678.0,-0.5,26,1,7,5,371.116,371.116,0,1,0,0,0,0,0,0,0,Between -11 and 5
+573,"(2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile",291.26,150.0,456.0,-1.4,21,4,8,2,291.097,291.097,0,1,0,4,4,0,0,0,0,Between -11 and 5
+574,Metformin glycinate,204.23,155.0,175.0,,14,5,4,3,204.133,204.133,0,2,0,0,0,0,0,0,0,Larger than 6
+575,1-Palmityl-2-(4-carboxybutyl)-SN-glycero-3-phosphocholine,581.8,114.0,606.0,6.7,39,1,8,30,581.406,581.406,0,1,0,1,1,0,0,0,0,Larger than 6
+576,3-phenyl-4-propyl-1-(pyridin-2-yl)-1H-pyrazol-5-ol,279.34,45.2,412.0,3.7,21,1,3,4,279.137,279.137,0,1,0,0,0,0,0,0,0,Between -11 and 5
+577,Ozanimod,404.5,104.0,609.0,3.1,30,2,7,7,404.185,404.185,0,1,0,1,1,0,0,0,0,Between -11 and 5
+578,Sabizabulin,377.4,89.2,534.0,3.4,28,2,5,6,377.138,377.138,0,1,0,0,0,0,0,0,0,Between -11 and 5
+579,Zavegepant,638.8,117.0,1160.0,3.1,47,3,6,6,638.369,638.369,0,1,0,1,1,0,0,0,0,Between -11 and 5
+580,CID 53477736,749.0,180.0,1150.0,4.0,52,5,14,7,748.509,748.509,0,1,0,18,18,0,0,0,0,Between -11 and 5
+581,Danoprevir (RG7227),731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,5,0,1,1,0,Between -11 and 5
+582,Solu-Medrol,497.5,138.0,981.0,,35,3,8,7,497.215,497.215,1,2,0,8,8,0,0,0,0,Larger than 6
+583,Vericiguat,426.4,147.0,622.0,1.5,31,3,10,5,426.136,426.136,0,1,0,0,0,0,0,0,0,Between -11 and 5
+584,Aldose reductase-IN-1,421.4,137.0,706.0,2.3,29,1,11,4,421.046,421.046,0,1,0,0,0,0,0,0,0,Between -11 and 5
+585,Ixazomib citrate,517.1,168.0,797.0,,34,4,9,11,516.087,516.087,0,1,0,1,1,0,0,0,0,Larger than 6
+586,Vidofludimus calcium anhydrous,748.8,157.0,571.0,,53,2,10,8,748.191,748.191,0,3,0,0,0,0,0,0,0,Larger than 6
+587,Upadacitinib,380.4,78.3,561.0,2.7,27,2,6,3,380.157,380.157,0,1,0,2,2,0,0,0,0,Between -11 and 5
+588,Asapiprant,501.6,131.0,789.0,3.1,35,1,10,9,501.157,501.157,0,1,0,0,0,0,0,0,0,Between -11 and 5
+589,"1,1'-hexamethylene bis[5-(p-chlorophenyl) biguanide] di-D-gluconate",735.7,317.0,819.0,,48,13,10,18,734.249,734.249,0,3,0,4,4,0,2,2,0,Larger than 6
+590,Proxalutamide,517.5,118.0,894.0,4.3,36,0,10,6,517.12,517.12,0,1,0,0,0,0,0,0,0,Between -11 and 5
+591,Rocefin,652.6,300.0,1120.0,,41,5,17,7,652.042,652.042,0,6,0,2,2,0,1,1,0,Larger than 6
+592,Galidesivir hydrochloride,301.73,140.0,334.0,,20,7,7,2,301.094,301.094,0,2,0,4,4,0,0,0,0,Larger than 6
+593,Entresto,1916.0,396.0,1140.0,,135,7,29,40,1915.81,1914.81,0,15,0,6,6,0,0,0,0,Larger than 6
+594,"(2R,6S,12Z,13aS,14aR,16aS)-6-[(tert-Butoxycarbonyl)amino]-14a-[(cyclopropylsulfonyl)carbamoyl]-5,16-dioxo-1,2,3,5,6,7,8,9,10,11,13a,14,14a,15,16,16a-hexadecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-2-yl 4-fluoro-1,3-dihydro-2H-isoindole-2-carboxylate",731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,5,0,1,1,0,Between -11 and 5
+595,Harvoni,1418.4,327.0,2730.0,,101,7,21,23,1417.58,1417.58,0,2,0,12,12,0,0,0,0,Larger than 6
+596,Abivertinib,487.5,98.4,752.0,4.2,36,3,8,7,487.213,487.213,0,1,0,0,0,0,0,0,0,Between -11 and 5
+597,Legalon SIL,1453.1,495.0,1200.0,,102,6,32,24,1452.23,1452.23,0,6,0,8,8,0,0,0,0,Larger than 6
+598,Maltofer,449.16,200.0,367.0,,27,11,14,8,449.059,449.059,0,5,0,9,9,0,0,0,0,Larger than 6
+599,Zunsemetinib,513.9,101.0,888.0,2.5,36,1,9,6,513.138,513.138,0,1,0,0,0,0,0,0,0,Between -11 and 5
+600,Treamid,318.37,116.0,345.0,-0.7,23,4,4,10,318.18,318.18,0,1,0,0,0,0,0,0,0,Between -11 and 5
+601,CID 87071853,1003.2,299.0,767.0,,67,6,20,20,1002.3,1002.3,0,3,0,4,4,0,2,2,0,Larger than 6
+602,"disodium;[2-[(8S,9R,10S,11S,13S,14S,16R,17R)-9-fluoro-11,17-dihydroxy-10,13,16-trimethyl-3-oxo-6,7,8,11,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl]-2-oxoethyl] phosphate;hydrate",534.4,148.0,962.0,,35,3,10,3,534.141,534.141,0,4,0,8,8,0,0,0,0,Larger than 6
+603,Heparin sodium,1157.9,652.0,2410.0,,71,15,38,21,1157.0,1157.0,1,2,0,20,0,20,0,0,0,Larger than 6
+604,CID 101731853,2088.6,933.0,4400.0,3.1,140,40,48,73,2086.96,2086.96,0,1,0,18,16,2,0,0,0,Between -11 and 5
+605,"N-(5-Oxidanyl-1,3-Benzothiazol-2-Yl)ethanamide",208.24,90.5,237.0,1.5,14,2,4,1,208.031,208.031,0,1,0,0,0,0,0,0,0,Between -11 and 5
+606,Danicopan,580.4,123.0,891.0,3.3,38,1,8,6,579.103,579.103,0,1,0,2,2,0,0,0,0,Between -11 and 5
+607,"Disodium;2-[[2-[[4-(2,2-dimethylpropanoyloxy)phenyl]sulfonylamino]benzoyl]amino]acetate",479.4,150.0,726.0,,32,2,8,8,479.086,479.086,1,3,0,0,0,0,0,0,0,Larger than 6
+608,"propan-2-yl (2S)-2-[[[(3R,4R,5R)-5-[2-amino-6-(methylamino)purin-9-yl]-4-fluoro-3-hydroxy-4-methyloxolan-2-yl]methoxy-phenoxyphosphoryl]amino]propanoate",581.5,185.0,919.0,1.7,40,4,14,12,581.216,581.216,0,1,0,6,5,1,0,0,0,Between -11 and 5
+609,"[(1S,4R,6S,7E,18R)-4-(cyclopropylsulfonylcarbamoyl)-14-[(2-methylpropan-2-yl)oxycarbonylamino]-2,15-dioxo-3,16-diazatricyclo[14.3.0.04,6]nonadec-7-en-18-yl] 4-fluoro-1,3-dihydroisoindole-2-carboxylate",731.8,189.0,1530.0,3.3,51,3,10,8,731.3,731.3,0,1,0,5,4,1,1,1,0,Between -11 and 5
+610,Eclitasertib,378.4,113.0,570.0,1.7,28,2,6,4,378.144,378.144,0,1,0,1,1,0,0,0,0,Between -11 and 5
+611,Dazcapistat,395.4,115.0,611.0,2.9,29,2,6,7,395.128,395.128,0,1,0,1,0,1,0,0,0,Between -11 and 5
+612,Bexotegrast,492.6,113.0,655.0,1.8,36,3,9,14,492.285,492.285,0,1,0,1,1,0,0,0,0,Between -11 and 5
+613,Estetrol monohydrate,322.4,81.9,441.0,,23,5,5,0,322.178,322.178,0,2,0,7,7,0,0,0,0,Larger than 6
+614,Sildenafil,474.6,118.0,838.0,1.5,33,1,8,7,474.205,474.205,0,1,0,0,0,0,0,0,0,Between -11 and 5
+615,Azilsartan,456.4,115.0,783.0,4.4,34,2,7,7,456.143,456.143,0,1,0,0,0,0,0,0,0,Between -11 and 5
+616,Echinochrome A,266.2,135.0,455.0,2.0,19,5,7,1,266.043,266.043,0,1,0,0,0,0,0,0,0,Between -11 and 5
+617,F-Arag F-18,284.23,135.0,449.0,-0.9,20,4,7,2,284.09,284.09,0,1,1,4,4,0,0,0,0,Between -11 and 5
+618,Apabetalone,370.4,89.4,543.0,2.3,27,2,6,6,370.153,370.153,0,1,0,0,0,0,0,0,0,Between -11 and 5
+619,"4-acetamidobenzoic acid;9-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1H-purin-6-one;(2R)-1-(dimethylamino)propan-2-ol",1115.2,399.0,658.0,,79,13,22,14,1114.55,1114.55,0,7,0,7,7,0,0,0,0,Larger than 6
+620,"[(1R,3R)-4-[(15Z,17E)-16-formyl-18-(4-hydroxy-2,2,6,6-tetramethylcyclohexyl)-3,7,12-trimethyl-14-oxooctadeca-2,4,6,8,10,12,15,17-octaenylidene]-3-hydroxy-3,5,5-trimethylcyclohexyl] acetate",672.9,101.0,1480.0,9.3,49,2,6,13,672.439,672.439,0,1,0,2,2,0,9,2,7,Larger than 6
+621,Abivertinib maleate,639.6,175.0,871.0,,46,7,14,9,639.245,639.245,0,4,0,0,0,0,1,1,0,Larger than 6
+622,"7-[[2-(2-Amino-1,3-thiazol-4-yl)-2-(2,2-dimethylpropanoyloxymethoxyimino)acetyl]amino]-3-ethenyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid",509.6,227.0,961.0,1.1,34,3,12,10,509.104,509.104,0,1,0,2,0,2,1,0,1,Between -11 and 5
+623,Unii-T5UX5skk2S,452.5,120.0,723.0,3.5,33,4,4,9,452.242,452.242,0,1,0,3,3,0,0,0,0,Between -11 and 5
+624,"azane;(2R,3S,4S,5R,6R)-2-(hydroxymethyl)-6-[(2R,3S,4R,5R,6S)-4,5,6-trihydroxy-2-(hydroxymethyl)oxan-3-yl]oxyoxane-3,4,5-triol",359.33,191.0,382.0,,24,9,12,4,359.143,359.143,0,2,0,10,10,0,0,0,0,Larger than 6
+625,Nezulcitinib,527.7,104.0,866.0,3.6,39,3,6,6,527.301,527.301,0,1,0,1,1,0,0,0,0,Between -11 and 5
+626,P9Zqs28F8C,403.5,86.9,712.0,3.6,28,2,4,4,403.193,403.193,0,1,0,1,1,0,1,1,0,Between -11 and 5
+627,Lufotrelvir,552.5,196.0,927.0,0.5,38,6,9,13,552.199,552.199,0,1,0,3,3,0,0,0,0,Between -11 and 5
+628,Nirmatrelvir,499.5,131.0,964.0,2.2,35,3,8,7,499.241,499.241,0,1,0,6,6,0,0,0,0,Between -11 and 5
+629,"hexasodium;4-[[(2S,4R)-5-ethoxy-4-methyl-5-oxo-1-(4-phenylphenyl)pentan-2-yl]amino]-4-oxobutanoate;hydride;(2S)-3-methyl-2-[pentanoyl-[[4-[2-(1,2,3-triaza-4-azanidacyclopenta-2,5-dien-5-yl)phenyl]phenyl]methyl]amino]butanoate;pentahydrate",1922.0,396.0,1140.0,,135,7,35,40,1921.86,1920.85,-6,21,0,6,6,0,0,0,0,Larger than 6
+630,Nangibotide,1342.5,634.0,2630.0,-8.3,92,20,24,45,1341.53,1341.53,0,1,0,10,10,0,0,0,0,Between -11 and 5
+
+
+
+
+
+
+ ]]>
+ Python
+ Shiny
+ Pandas
+ Plotly
+ PubChem
+ Cheminformatics
+ https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_app_embed.html
+ Sun, 07 May 2023 12:00:00 GMT
+
+
+ Shinylive app in Python
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_data_prep.html
+
+
Brief introduction
+
Since I’ve had a lot of fun building a Shiny app in R last time, I was on track to build another Shiny app again but using Python instead. So here in this post, I’ll talk about the data wrangling process to prepare the final dataset needed to build a Shinylive app in Python. The actual Shinylive app deployment and access will be shown in a separate post after this one.
+
+
+
+
Source of data
+
The dataset used for this Shiny app in Python was from PubChem (link here). There were a total of 631 compounds at the time when I downloaded them as .csv file, along with their relevant compound data. I only picked this dataset randomly, as the focus would be more on app building, but it was nice to see an interactive web app being built and used for a domain such as pharmaceutical research.
+
+
+
+
Import Polars
+
Polars dataframe library was used again this time.
+
+
import polars as pl
+
+
+
+
+
Reading .csv file
+
+
pc = pl.read_csv("pubchem.csv")
+pc.head()
+
+
+
+
+shape: (5, 38)
+
+
+
+
+cid
+
+
+cmpdname
+
+
+cmpdsynonym
+
+
+mw
+
+
+mf
+
+
+polararea
+
+
+complexity
+
+
+xlogp
+
+
+heavycnt
+
+
+hbonddonor
+
+
+hbondacc
+
+
+rotbonds
+
+
+inchi
+
+
+isosmiles
+
+
+canonicalsmiles
+
+
+inchikey
+
+
+iupacname
+
+
+exactmass
+
+
+monoisotopicmass
+
+
+charge
+
+
+covalentunitcnt
+
+
+isotopeatomcnt
+
+
+totalatomstereocnt
+
+
+definedatomstereocnt
+
+
+undefinedatomstereocnt
+
+
+totalbondstereocnt
+
+
+definedbondstereocnt
+
+
+undefinedbondstereocnt
+
+
+pclidcnt
+
+
+gpidcnt
+
+
+meshheadings
+
+
+annothits
+
+
+annothitcnt
+
+
+aids
+
+
+cidcdate
+
+
+sidsrcname
+
+
+depcatg
+
+
+annotation
+
+
+
+
+i64
+
+
+str
+
+
+str
+
+
+f64
+
+
+str
+
+
+f64
+
+
+f64
+
+
+str
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+f64
+
+
+f64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+str
+
+
+str
+
+
+i64
+
+
+str
+
+
+i64
+
+
+str
+
+
+str
+
+
+str
+
+
+
+
+
+
+5280453
+
+
+"Calcitriol"
+
+
+"calcitriol|322...
+
+
+416.6
+
+
+"C27H44O3"
+
+
+60.7
+
+
+688.0
+
+
+"5.100"
+
+
+30
+
+
+3
+
+
+3
+
+
+6
+
+
+"InChI=1S/C27H4...
+
+
+"C[C@H](CCCC(C)...
+
+
+"CC(CCCC(C)(C)O...
+
+
+"GMRQFYUYWCNGIN...
+
+
+"(1R,3S,5Z)-5-[...
+
+
+416.329
+
+
+416.329
+
+
+0
+
+
+1
+
+
+0
+
+
+6
+
+
+6
+
+
+0
+
+
+2
+
+
+2
+
+
+0
+
+
+22311
+
+
+46029
+
+
+"Calcitriol"
+
+
+"Biological Tes...
+
+
+12
+
+
+"485|631|731|78...
+
+
+20040916
+
+
+"A2B Chem|AA BL...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+9962735
+
+
+"Ubiquinol"
+
+
+"ubiquinol|992-...
+
+
+865.4
+
+
+"C59H92O4"
+
+
+58.9
+
+
+1600.0
+
+
+"20.200"
+
+
+63
+
+
+2
+
+
+4
+
+
+31
+
+
+"InChI=1S/C59H9...
+
+
+"CC1=C(C(=C(C(=...
+
+
+"CC1=C(C(=C(C(=...
+
+
+"QNTNKSLOFHEFPK...
+
+
+"2-[(2E,6E,10E,...
+
+
+864.7
+
+
+864.7
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+9
+
+
+9
+
+
+0
+
+
+2732
+
+
+21358
+
+
+"NULL"
+
+
+"Chemical and P...
+
+
+7
+
+
+"NULL"
+
+
+20061025
+
+
+"001Chemical|A2...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+5961
+
+
+"Glutamine"
+
+
+"L-glutamine|gl...
+
+
+146.14
+
+
+"C5H10N2O3"
+
+
+106.0
+
+
+146.0
+
+
+"-3.100"
+
+
+10
+
+
+3
+
+
+4
+
+
+4
+
+
+"InChI=1S/C5H10...
+
+
+"C(CC(=O)N)[C@@...
+
+
+"C(CC(=O)N)C(C(...
+
+
+"ZDXPYRJPNDTMRX...
+
+
+"(2S)-2,5-diami...
+
+
+146.069
+
+
+146.069
+
+
+0
+
+
+1
+
+
+0
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+88218
+
+
+399
+
+
+"Glutamine"
+
+
+"Biological Tes...
+
+
+12
+
+
+"422|429|436|54...
+
+
+20040916
+
+
+"001Chemical|3B...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+2244
+
+
+"Aspirin"
+
+
+"aspirin|ACETYL...
+
+
+180.16
+
+
+"C9H8O4"
+
+
+63.6
+
+
+212.0
+
+
+"1.200"
+
+
+13
+
+
+1
+
+
+4
+
+
+3
+
+
+"InChI=1S/C9H8O...
+
+
+"CC(=O)OC1=CC=C...
+
+
+"CC(=O)OC1=CC=C...
+
+
+"BSYNRYMUTXBXSQ...
+
+
+"2-acetyloxyben...
+
+
+180.042
+
+
+180.042
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+127012
+
+
+364455
+
+
+"Aspirin"
+
+
+"Biological Tes...
+
+
+12
+
+
+"1|3|9|15|19|21...
+
+
+20040916
+
+
+"001Chemical|3B...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+457
+
+
+"1-Methylnicoti...
+
+
+"1-methylnicoti...
+
+
+137.16
+
+
+"C7H9N2O+"
+
+
+47.0
+
+
+136.0
+
+
+"-0.100"
+
+
+10
+
+
+1
+
+
+1
+
+
+1
+
+
+"InChI=1S/C7H8N...
+
+
+"C[N+]1=CC=CC(=...
+
+
+"C[N+]1=CC=CC(=...
+
+
+"LDHMAVIPBRSVRG...
+
+
+"1-methylpyridi...
+
+
+137.071
+
+
+137.071
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+310
+
+
+674
+
+
+"NULL"
+
+
+"Biological Tes...
+
+
+8
+
+
+"61001|61002|14...
+
+
+20040916
+
+
+"001Chemical|3B...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+
+
+
+
+
+
+
Quick look at the data
+
I decided to comment out the code below to keep the post at a reasonable length for reading purpose, but they were very handy for a quick glimpse of the data content.
+
+
# Quick overview of the variables in each column in the dataset
+# Uncomment line below if needed to run
+#print(pc.glimpse())
+
+# Quick look at all column names
+# Uncomment line below if needed to run
+#pc.columns
+
+
+
+
+
Check for nulls in dataset
+
+
pc.null_count()
+
+
+
+
+shape: (1, 38)
+
+
+
+
+cid
+
+
+cmpdname
+
+
+cmpdsynonym
+
+
+mw
+
+
+mf
+
+
+polararea
+
+
+complexity
+
+
+xlogp
+
+
+heavycnt
+
+
+hbonddonor
+
+
+hbondacc
+
+
+rotbonds
+
+
+inchi
+
+
+isosmiles
+
+
+canonicalsmiles
+
+
+inchikey
+
+
+iupacname
+
+
+exactmass
+
+
+monoisotopicmass
+
+
+charge
+
+
+covalentunitcnt
+
+
+isotopeatomcnt
+
+
+totalatomstereocnt
+
+
+definedatomstereocnt
+
+
+undefinedatomstereocnt
+
+
+totalbondstereocnt
+
+
+definedbondstereocnt
+
+
+undefinedbondstereocnt
+
+
+pclidcnt
+
+
+gpidcnt
+
+
+meshheadings
+
+
+annothits
+
+
+annothitcnt
+
+
+aids
+
+
+cidcdate
+
+
+sidsrcname
+
+
+depcatg
+
+
+annotation
+
+
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+u32
+
+
+
+
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+
+
+
+
+
+
+
+
+
Change column names as needed
+
+
# Change column names
+pc_cov = pc.rename(
+ {
+"cmpdname": "Compound name",
+"cmpdsynonym": "Synonyms",
+"mw": "Molecular weight",
+"mf": "Molecular formula",
+"polararea": "Polar surface area",
+"complexity": "Complexity",
+"xlogp": "Partition coefficients",
+"heavycnt": "Heavy atom count",
+"hbonddonor": "Hydrogen bond donor count",
+"hbondacc": "Hydrogen bond acceptor count",
+"rotbonds": "Rotatable bond count",
+"exactmass": "Exact mass",
+"monoisotopicmass": "Monoisotopic mass",
+"charge": "Formal charge",
+"covalentunitcnt": "Covalently-bonded unit count",
+"isotopeatomcnt": "Isotope atom count",
+"totalatomstereocnt": "Total atom stereocenter count",
+"definedatomstereocnt": "Defined atom stereocenter count",
+"undefinedatomstereocnt": "Undefined atoms stereocenter count",
+"totalbondstereocnt": "Total bond stereocenter count",
+"definedbondstereocnt": "Defined bond stereocenter count",
+"undefinedbondstereocnt": "Undefined bond stereocenter count",
+"meshheadings": "MeSH headings"
+ }
+)
+
+pc_cov.head()
+
+
+
+
+shape: (5, 38)
+
+
+
+
+cid
+
+
+Compound name
+
+
+Synonyms
+
+
+Molecular weight
+
+
+Molecular formula
+
+
+Polar surface area
+
+
+Complexity
+
+
+Partition coefficients
+
+
+Heavy atom count
+
+
+Hydrogen bond donor count
+
+
+Hydrogen bond acceptor count
+
+
+Rotatable bond count
+
+
+inchi
+
+
+isosmiles
+
+
+canonicalsmiles
+
+
+inchikey
+
+
+iupacname
+
+
+Exact mass
+
+
+Monoisotopic mass
+
+
+Formal charge
+
+
+Covalently-bonded unit count
+
+
+Isotope atom count
+
+
+Total atom stereocenter count
+
+
+Defined atom stereocenter count
+
+
+Undefined atoms stereocenter count
+
+
+Total bond stereocenter count
+
+
+Defined bond stereocenter count
+
+
+Undefined bond stereocenter count
+
+
+pclidcnt
+
+
+gpidcnt
+
+
+MeSH headings
+
+
+annothits
+
+
+annothitcnt
+
+
+aids
+
+
+cidcdate
+
+
+sidsrcname
+
+
+depcatg
+
+
+annotation
+
+
+
+
+i64
+
+
+str
+
+
+str
+
+
+f64
+
+
+str
+
+
+f64
+
+
+f64
+
+
+str
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+f64
+
+
+f64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+str
+
+
+str
+
+
+i64
+
+
+str
+
+
+i64
+
+
+str
+
+
+str
+
+
+str
+
+
+
+
+
+
+5280453
+
+
+"Calcitriol"
+
+
+"calcitriol|322...
+
+
+416.6
+
+
+"C27H44O3"
+
+
+60.7
+
+
+688.0
+
+
+"5.100"
+
+
+30
+
+
+3
+
+
+3
+
+
+6
+
+
+"InChI=1S/C27H4...
+
+
+"C[C@H](CCCC(C)...
+
+
+"CC(CCCC(C)(C)O...
+
+
+"GMRQFYUYWCNGIN...
+
+
+"(1R,3S,5Z)-5-[...
+
+
+416.329
+
+
+416.329
+
+
+0
+
+
+1
+
+
+0
+
+
+6
+
+
+6
+
+
+0
+
+
+2
+
+
+2
+
+
+0
+
+
+22311
+
+
+46029
+
+
+"Calcitriol"
+
+
+"Biological Tes...
+
+
+12
+
+
+"485|631|731|78...
+
+
+20040916
+
+
+"A2B Chem|AA BL...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+9962735
+
+
+"Ubiquinol"
+
+
+"ubiquinol|992-...
+
+
+865.4
+
+
+"C59H92O4"
+
+
+58.9
+
+
+1600.0
+
+
+"20.200"
+
+
+63
+
+
+2
+
+
+4
+
+
+31
+
+
+"InChI=1S/C59H9...
+
+
+"CC1=C(C(=C(C(=...
+
+
+"CC1=C(C(=C(C(=...
+
+
+"QNTNKSLOFHEFPK...
+
+
+"2-[(2E,6E,10E,...
+
+
+864.7
+
+
+864.7
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+9
+
+
+9
+
+
+0
+
+
+2732
+
+
+21358
+
+
+"NULL"
+
+
+"Chemical and P...
+
+
+7
+
+
+"NULL"
+
+
+20061025
+
+
+"001Chemical|A2...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+5961
+
+
+"Glutamine"
+
+
+"L-glutamine|gl...
+
+
+146.14
+
+
+"C5H10N2O3"
+
+
+106.0
+
+
+146.0
+
+
+"-3.100"
+
+
+10
+
+
+3
+
+
+4
+
+
+4
+
+
+"InChI=1S/C5H10...
+
+
+"C(CC(=O)N)[C@@...
+
+
+"C(CC(=O)N)C(C(...
+
+
+"ZDXPYRJPNDTMRX...
+
+
+"(2S)-2,5-diami...
+
+
+146.069
+
+
+146.069
+
+
+0
+
+
+1
+
+
+0
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+88218
+
+
+399
+
+
+"Glutamine"
+
+
+"Biological Tes...
+
+
+12
+
+
+"422|429|436|54...
+
+
+20040916
+
+
+"001Chemical|3B...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+2244
+
+
+"Aspirin"
+
+
+"aspirin|ACETYL...
+
+
+180.16
+
+
+"C9H8O4"
+
+
+63.6
+
+
+212.0
+
+
+"1.200"
+
+
+13
+
+
+1
+
+
+4
+
+
+3
+
+
+"InChI=1S/C9H8O...
+
+
+"CC(=O)OC1=CC=C...
+
+
+"CC(=O)OC1=CC=C...
+
+
+"BSYNRYMUTXBXSQ...
+
+
+"2-acetyloxyben...
+
+
+180.042
+
+
+180.042
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+127012
+
+
+364455
+
+
+"Aspirin"
+
+
+"Biological Tes...
+
+
+12
+
+
+"1|3|9|15|19|21...
+
+
+20040916
+
+
+"001Chemical|3B...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+457
+
+
+"1-Methylnicoti...
+
+
+"1-methylnicoti...
+
+
+137.16
+
+
+"C7H9N2O+"
+
+
+47.0
+
+
+136.0
+
+
+"-0.100"
+
+
+10
+
+
+1
+
+
+1
+
+
+1
+
+
+"InChI=1S/C7H8N...
+
+
+"C[N+]1=CC=CC(=...
+
+
+"C[N+]1=CC=CC(=...
+
+
+"LDHMAVIPBRSVRG...
+
+
+"1-methylpyridi...
+
+
+137.071
+
+
+137.071
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+310
+
+
+674
+
+
+"NULL"
+
+
+"Biological Tes...
+
+
+8
+
+
+"61001|61002|14...
+
+
+20040916
+
+
+"001Chemical|3B...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+
+
+
+
+
+
+
Definitions of molecular properties in this PubChem dataset
+
The definitions for some of the column names were shown below, which were mainly derived and adapted from PubChem:
+
Note: please refer to PubChem documentations for full definitions
+
+
Molecular weight - molecular mass of compounds measured in daltons
+
Topological polar surface area - measured as an estimate of polar surface area of a molecule (i.e. the surface sum over polar atoms in a molecule), with units in angstrom squared (Å2)
+
Complexity - complexity rating for compounds, based on Bertz/Hendrickson/Ihlenfeldt formula as a rough estimation of how complex a compound was structurally
+
Partition coefficients (xlogp) - predicted octanol-water partition coefficient as a measure of the hydrophilicity or hydrophobicity of a molecule
+
Heavy atom count - number of heavy atoms e.g. non-hydrogen atoms in the compound
+
Hydrogen bond donor count - number of hydrogen bond donors in the compound
+
Hydrogen bond acceptor count - number of hydrogen bond acceptors in the compound
+
Rotatable bond count - defined as any single-order non-ring bond, where atoms on either side of the bond were in turn bound to non-terminal heavy atoms (e.g. non-hydrogen). Rotation around the bond axis would change overall molecule shape and generate conformers which could be distinguished by standard spectroscopic methods
+
Exact mass - exact mass of an isotopic species, obtained by summing masses of individual isotopes of the molecule
+
Monoisotopic mass - sum of the masses of atoms in a molecule, using unbound, ground-state, rest mass of principal (or most abundant) isotope for each element instead of isotopic average mass
+
Formal charge - the difference between the number of valence electrons of each atom, and the number of electrons the atom was associated with, assumed any shared electrons were equally shared between the two bonded atoms
+
Covalently-bonded unit count - a group of atoms connected by covalent bonds, ignoring other bond types (or a single atom without covalent bonds), representing number of such units in the compound
+
Isotope atom count - number of isotopes that were not most abundant for the corresponding chemical elements. Isotopes were variants of a chemical element that differed in neutron number
+
Defined atom stereocenter count - atom stereocenter (or chiral center) was where an atom was attached to 4 different types of atoms or groups of atoms in a tetrahedral arrangement. It could either be (R)- or (S)- configurations. Some of the compounds e.g. racemic mixtures, could have undefined atom stereocenter, where (R/S)-config was not specifically defined. Defined atom stereocenter count was the number of atom stereocenters where configurations were specifically defined
+
Undefined atoms stereocenter count - this was the undefined version of the atoms stereocenter count
+
Defined bond stereocenter count - bond stereocenter (or non-rotatable bond) was where two atoms could have different arrangement e.g. in cis- & trans- forms of butene around its double bond. Some compounds could have an undefined bond stereocenter (stereochemistry not specifically defined). Defined bond stereocenter count was the number of bond stereocenters where configurations were specifically defined.
+
Undefined bond stereocenter count - this was the undefined version of the bond stereocenter count
+
+
+
+
+
Convert data type for selected columns
+
+
# Convert data type - only for partition coefficients column (rest were okay)
+pc_cov = pc_cov.with_column((pl.col("Partition coefficients")).cast(pl.Float64, strict =False))
+pc_cov.head()
+
+
+
+
+shape: (5, 38)
+
+
+
+
+cid
+
+
+Compound name
+
+
+Synonyms
+
+
+Molecular weight
+
+
+Molecular formula
+
+
+Polar surface area
+
+
+Complexity
+
+
+Partition coefficients
+
+
+Heavy atom count
+
+
+Hydrogen bond donor count
+
+
+Hydrogen bond acceptor count
+
+
+Rotatable bond count
+
+
+inchi
+
+
+isosmiles
+
+
+canonicalsmiles
+
+
+inchikey
+
+
+iupacname
+
+
+Exact mass
+
+
+Monoisotopic mass
+
+
+Formal charge
+
+
+Covalently-bonded unit count
+
+
+Isotope atom count
+
+
+Total atom stereocenter count
+
+
+Defined atom stereocenter count
+
+
+Undefined atoms stereocenter count
+
+
+Total bond stereocenter count
+
+
+Defined bond stereocenter count
+
+
+Undefined bond stereocenter count
+
+
+pclidcnt
+
+
+gpidcnt
+
+
+MeSH headings
+
+
+annothits
+
+
+annothitcnt
+
+
+aids
+
+
+cidcdate
+
+
+sidsrcname
+
+
+depcatg
+
+
+annotation
+
+
+
+
+i64
+
+
+str
+
+
+str
+
+
+f64
+
+
+str
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+str
+
+
+f64
+
+
+f64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+str
+
+
+str
+
+
+i64
+
+
+str
+
+
+i64
+
+
+str
+
+
+str
+
+
+str
+
+
+
+
+
+
+5280453
+
+
+"Calcitriol"
+
+
+"calcitriol|322...
+
+
+416.6
+
+
+"C27H44O3"
+
+
+60.7
+
+
+688.0
+
+
+5.1
+
+
+30
+
+
+3
+
+
+3
+
+
+6
+
+
+"InChI=1S/C27H4...
+
+
+"C[C@H](CCCC(C)...
+
+
+"CC(CCCC(C)(C)O...
+
+
+"GMRQFYUYWCNGIN...
+
+
+"(1R,3S,5Z)-5-[...
+
+
+416.329
+
+
+416.329
+
+
+0
+
+
+1
+
+
+0
+
+
+6
+
+
+6
+
+
+0
+
+
+2
+
+
+2
+
+
+0
+
+
+22311
+
+
+46029
+
+
+"Calcitriol"
+
+
+"Biological Tes...
+
+
+12
+
+
+"485|631|731|78...
+
+
+20040916
+
+
+"A2B Chem|AA BL...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+9962735
+
+
+"Ubiquinol"
+
+
+"ubiquinol|992-...
+
+
+865.4
+
+
+"C59H92O4"
+
+
+58.9
+
+
+1600.0
+
+
+20.2
+
+
+63
+
+
+2
+
+
+4
+
+
+31
+
+
+"InChI=1S/C59H9...
+
+
+"CC1=C(C(=C(C(=...
+
+
+"CC1=C(C(=C(C(=...
+
+
+"QNTNKSLOFHEFPK...
+
+
+"2-[(2E,6E,10E,...
+
+
+864.7
+
+
+864.7
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+9
+
+
+9
+
+
+0
+
+
+2732
+
+
+21358
+
+
+"NULL"
+
+
+"Chemical and P...
+
+
+7
+
+
+"NULL"
+
+
+20061025
+
+
+"001Chemical|A2...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+5961
+
+
+"Glutamine"
+
+
+"L-glutamine|gl...
+
+
+146.14
+
+
+"C5H10N2O3"
+
+
+106.0
+
+
+146.0
+
+
+-3.1
+
+
+10
+
+
+3
+
+
+4
+
+
+4
+
+
+"InChI=1S/C5H10...
+
+
+"C(CC(=O)N)[C@@...
+
+
+"C(CC(=O)N)C(C(...
+
+
+"ZDXPYRJPNDTMRX...
+
+
+"(2S)-2,5-diami...
+
+
+146.069
+
+
+146.069
+
+
+0
+
+
+1
+
+
+0
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+88218
+
+
+399
+
+
+"Glutamine"
+
+
+"Biological Tes...
+
+
+12
+
+
+"422|429|436|54...
+
+
+20040916
+
+
+"001Chemical|3B...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+2244
+
+
+"Aspirin"
+
+
+"aspirin|ACETYL...
+
+
+180.16
+
+
+"C9H8O4"
+
+
+63.6
+
+
+212.0
+
+
+1.2
+
+
+13
+
+
+1
+
+
+4
+
+
+3
+
+
+"InChI=1S/C9H8O...
+
+
+"CC(=O)OC1=CC=C...
+
+
+"CC(=O)OC1=CC=C...
+
+
+"BSYNRYMUTXBXSQ...
+
+
+"2-acetyloxyben...
+
+
+180.042
+
+
+180.042
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+127012
+
+
+364455
+
+
+"Aspirin"
+
+
+"Biological Tes...
+
+
+12
+
+
+"1|3|9|15|19|21...
+
+
+20040916
+
+
+"001Chemical|3B...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+457
+
+
+"1-Methylnicoti...
+
+
+"1-methylnicoti...
+
+
+137.16
+
+
+"C7H9N2O+"
+
+
+47.0
+
+
+136.0
+
+
+-0.1
+
+
+10
+
+
+1
+
+
+1
+
+
+1
+
+
+"InChI=1S/C7H8N...
+
+
+"C[N+]1=CC=CC(=...
+
+
+"C[N+]1=CC=CC(=...
+
+
+"LDHMAVIPBRSVRG...
+
+
+"1-methylpyridi...
+
+
+137.071
+
+
+137.071
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+310
+
+
+674
+
+
+"NULL"
+
+
+"Biological Tes...
+
+
+8
+
+
+"61001|61002|14...
+
+
+20040916
+
+
+"001Chemical|3B...
+
+
+"Chemical Vendo...
+
+
+"COVID-19, COVI...
+
+
+
+
+
+
+
+
+
+
+
Select columns for data visualisations
+
The idea was really only keeping all the numerical columns for some data visualisations later. So I’ve dropped all the other columns in texts or of the string types.
+
+
# Drop unused columns in preparation for data visualisations
+pc_cov = pc_cov.drop([
+"cid",
+"Synonyms",
+"Molecular formula",
+"inchi",
+"isosmiles",
+"canonicalsmiles",
+"inchikey",
+"iupacname",
+"pclidcnt",
+"gpidcnt",
+"MeSH headings",
+"annothits",
+"annothitcnt",
+"aids",
+"cidcdate",
+"sidsrcname",
+"depcatg",
+"annotation"
+])
+
+pc_cov.head()
+
+
+
+
+shape: (5, 20)
+
+
+
+
+Compound name
+
+
+Molecular weight
+
+
+Polar surface area
+
+
+Complexity
+
+
+Partition coefficients
+
+
+Heavy atom count
+
+
+Hydrogen bond donor count
+
+
+Hydrogen bond acceptor count
+
+
+Rotatable bond count
+
+
+Exact mass
+
+
+Monoisotopic mass
+
+
+Formal charge
+
+
+Covalently-bonded unit count
+
+
+Isotope atom count
+
+
+Total atom stereocenter count
+
+
+Defined atom stereocenter count
+
+
+Undefined atoms stereocenter count
+
+
+Total bond stereocenter count
+
+
+Defined bond stereocenter count
+
+
+Undefined bond stereocenter count
+
+
+
+
+str
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+f64
+
+
+f64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+
+
+
+
+"Calcitriol"
+
+
+416.6
+
+
+60.7
+
+
+688.0
+
+
+5.1
+
+
+30
+
+
+3
+
+
+3
+
+
+6
+
+
+416.329
+
+
+416.329
+
+
+0
+
+
+1
+
+
+0
+
+
+6
+
+
+6
+
+
+0
+
+
+2
+
+
+2
+
+
+0
+
+
+
+
+"Ubiquinol"
+
+
+865.4
+
+
+58.9
+
+
+1600.0
+
+
+20.2
+
+
+63
+
+
+2
+
+
+4
+
+
+31
+
+
+864.7
+
+
+864.7
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+9
+
+
+9
+
+
+0
+
+
+
+
+"Glutamine"
+
+
+146.14
+
+
+106.0
+
+
+146.0
+
+
+-3.1
+
+
+10
+
+
+3
+
+
+4
+
+
+4
+
+
+146.069
+
+
+146.069
+
+
+0
+
+
+1
+
+
+0
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+
+
+"Aspirin"
+
+
+180.16
+
+
+63.6
+
+
+212.0
+
+
+1.2
+
+
+13
+
+
+1
+
+
+4
+
+
+3
+
+
+180.042
+
+
+180.042
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+
+
+"1-Methylnicoti...
+
+
+137.16
+
+
+47.0
+
+
+136.0
+
+
+-0.1
+
+
+10
+
+
+1
+
+
+1
+
+
+1
+
+
+137.071
+
+
+137.071
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+
+
+
+
+
+
+
+
+
Quick summary statistics of columns
+
+
# Overall descriptive statistics of kept columns
+pc_cov.describe()
+
+
+
+
+shape: (7, 21)
+
+
+
+
+describe
+
+
+Compound name
+
+
+Molecular weight
+
+
+Polar surface area
+
+
+Complexity
+
+
+Partition coefficients
+
+
+Heavy atom count
+
+
+Hydrogen bond donor count
+
+
+Hydrogen bond acceptor count
+
+
+Rotatable bond count
+
+
+Exact mass
+
+
+Monoisotopic mass
+
+
+Formal charge
+
+
+Covalently-bonded unit count
+
+
+Isotope atom count
+
+
+Total atom stereocenter count
+
+
+Defined atom stereocenter count
+
+
+Undefined atoms stereocenter count
+
+
+Total bond stereocenter count
+
+
+Defined bond stereocenter count
+
+
+Undefined bond stereocenter count
+
+
+
+
+str
+
+
+str
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+
+
+
+
+"count"
+
+
+"631"
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+631.0
+
+
+
+
+"null_count"
+
+
+"0"
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+173.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+
+
+"mean"
+
+
+null
+
+
+549.539675
+
+
+163.915368
+
+
+864.755626
+
+
+2.25917
+
+
+37.770206
+
+
+4.066561
+
+
+9.210777
+
+
+9.518225
+
+
+549.095022
+
+
+549.06013
+
+
+-0.004754
+
+
+1.578447
+
+
+0.006339
+
+
+4.017433
+
+
+3.551506
+
+
+0.465927
+
+
+0.381933
+
+
+0.343899
+
+
+0.038035
+
+
+
+
+"std"
+
+
+null
+
+
+455.236826
+
+
+192.256415
+
+
+1000.220379
+
+
+3.926459
+
+
+31.821967
+
+
+6.348004
+
+
+8.694184
+
+
+15.393131
+
+
+455.064211
+
+
+454.958033
+
+
+0.358537
+
+
+1.610416
+
+
+0.079429
+
+
+6.128363
+
+
+5.787792
+
+
+2.364089
+
+
+1.181171
+
+
+1.107245
+
+
+0.363159
+
+
+
+
+"min"
+
+
+"(+)-Mefloquine...
+
+
+103.1
+
+
+0.0
+
+
+0.0
+
+
+-24.0
+
+
+1.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+103.04
+
+
+103.04
+
+
+-6.0
+
+
+1.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+
+
+"max"
+
+
+"sodium;8-amino...
+
+
+4114.0
+
+
+1650.0
+
+
+9590.0
+
+
+20.2
+
+
+291.0
+
+
+57.0
+
+
+65.0
+
+
+151.0
+
+
+4112.12
+
+
+4111.12
+
+
+2.0
+
+
+21.0
+
+
+1.0
+
+
+39.0
+
+
+39.0
+
+
+31.0
+
+
+11.0
+
+
+11.0
+
+
+7.0
+
+
+
+
+"median"
+
+
+null
+
+
+435.9
+
+
+110.0
+
+
+635.0
+
+
+2.5
+
+
+30.0
+
+
+3.0
+
+
+7.0
+
+
+6.0
+
+
+435.227
+
+
+435.227
+
+
+0.0
+
+
+1.0
+
+
+0.0
+
+
+1.0
+
+
+1.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+0.0
+
+
+
+
+
+
+
+
+
+
+
Conditional assignments in Polars
+
The longer I’ve used Polars, the more I like its coding styles of chaining a string of different code functions together to manipulate dataframes in one go. This usually might mean that we could avoid writing some repeated loop functions to achieve the same results. In the example below, I’d like to show how to chain “when-then-otherwise” expressions by using Polars.
+
+
+
Chaining when-then-otherwise expressions - creating groups in data
+
I had the idea of separating all data into 3 different ranges of partition coefficients, so that this could be shown visually in plots. One of the possible ways (other than writing a loop function), or really the long way, to do this might be like the code shown below:
A shorter and probably more elegant way was to use the “when-then-otherwise” expression in Polars for conditional assignments (the following code snippet was adapted with thanks to the author of Polars, Ritchie Vink and also the good old Stack Overflow):
+
+
pc_cov = pc_cov.with_column(
+ pl.when((pl.col("Partition coefficients") <=-10))
+ .then("Smaller than -10")
+ .when((pl.col("Partition coefficients") >=-11) & (pl.col("Partition coefficients") <=5))
+ .then("Between -11 and 5")
+ .otherwise("Larger than 6")
+ .alias("Part_coef_group")
+)
+
+pc_cov.head(10)
+
+# a new column would be added to the end of the dataframe
+# with a new column name, "Part_coef_group"
+# (scroll to the very right to see the added column)
+
+
+
+
+shape: (10, 21)
+
+
+
+
+Compound name
+
+
+Molecular weight
+
+
+Polar surface area
+
+
+Complexity
+
+
+Partition coefficients
+
+
+Heavy atom count
+
+
+Hydrogen bond donor count
+
+
+Hydrogen bond acceptor count
+
+
+Rotatable bond count
+
+
+Exact mass
+
+
+Monoisotopic mass
+
+
+Formal charge
+
+
+Covalently-bonded unit count
+
+
+Isotope atom count
+
+
+Total atom stereocenter count
+
+
+Defined atom stereocenter count
+
+
+Undefined atoms stereocenter count
+
+
+Total bond stereocenter count
+
+
+Defined bond stereocenter count
+
+
+Undefined bond stereocenter count
+
+
+Part_coef_group
+
+
+
+
+str
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+f64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+f64
+
+
+f64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+i64
+
+
+str
+
+
+
+
+
+
+"Calcitriol"
+
+
+416.6
+
+
+60.7
+
+
+688.0
+
+
+5.1
+
+
+30
+
+
+3
+
+
+3
+
+
+6
+
+
+416.329
+
+
+416.329
+
+
+0
+
+
+1
+
+
+0
+
+
+6
+
+
+6
+
+
+0
+
+
+2
+
+
+2
+
+
+0
+
+
+"Larger than 6"
+
+
+
+
+"Ubiquinol"
+
+
+865.4
+
+
+58.9
+
+
+1600.0
+
+
+20.2
+
+
+63
+
+
+2
+
+
+4
+
+
+31
+
+
+864.7
+
+
+864.7
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+9
+
+
+9
+
+
+0
+
+
+"Larger than 6"
+
+
+
+
+"Glutamine"
+
+
+146.14
+
+
+106.0
+
+
+146.0
+
+
+-3.1
+
+
+10
+
+
+3
+
+
+4
+
+
+4
+
+
+146.069
+
+
+146.069
+
+
+0
+
+
+1
+
+
+0
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+"Between -11 an...
+
+
+
+
+"Aspirin"
+
+
+180.16
+
+
+63.6
+
+
+212.0
+
+
+1.2
+
+
+13
+
+
+1
+
+
+4
+
+
+3
+
+
+180.042
+
+
+180.042
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+"Between -11 an...
+
+
+
+
+"1-Methylnicoti...
+
+
+137.16
+
+
+47.0
+
+
+136.0
+
+
+-0.1
+
+
+10
+
+
+1
+
+
+1
+
+
+1
+
+
+137.071
+
+
+137.071
+
+
+1
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+"Between -11 an...
+
+
+
+
+"Losartan"
+
+
+422.9
+
+
+92.5
+
+
+520.0
+
+
+4.3
+
+
+30
+
+
+2
+
+
+5
+
+
+8
+
+
+422.162
+
+
+422.162
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+"Between -11 an...
+
+
+
+
+"Vitamin E"
+
+
+430.7
+
+
+29.5
+
+
+503.0
+
+
+10.7
+
+
+31
+
+
+1
+
+
+2
+
+
+12
+
+
+430.381
+
+
+430.381
+
+
+0
+
+
+1
+
+
+0
+
+
+3
+
+
+3
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+"Larger than 6"
+
+
+
+
+"Nicotinamide"
+
+
+122.12
+
+
+56.0
+
+
+114.0
+
+
+-0.4
+
+
+9
+
+
+1
+
+
+2
+
+
+1
+
+
+122.048
+
+
+122.048
+
+
+0
+
+
+1
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+"Between -11 an...
+
+
+
+
+"Adenosine"
+
+
+267.24
+
+
+140.0
+
+
+335.0
+
+
+-1.1
+
+
+19
+
+
+4
+
+
+8
+
+
+2
+
+
+267.097
+
+
+267.097
+
+
+0
+
+
+1
+
+
+0
+
+
+4
+
+
+4
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+"Between -11 an...
+
+
+
+
+"Inosine"
+
+
+268.23
+
+
+129.0
+
+
+405.0
+
+
+-1.3
+
+
+19
+
+
+4
+
+
+7
+
+
+2
+
+
+268.081
+
+
+268.081
+
+
+0
+
+
+1
+
+
+0
+
+
+4
+
+
+4
+
+
+0
+
+
+0
+
+
+0
+
+
+0
+
+
+"Between -11 an...
+
+
+
+
+
+
+
+
+
+
+
+
Import Plotly
+
Time for some data vizzes - importing Plotly first.
+
+
import plotly.express as px
+
+
+
+
+
Some examples of data visualisations
+
Below were some of the examples of building plots by using Plotly.
+
+
Partition coefficients vs. Molecular weights
+
+
fig = px.scatter(x = pc_cov["Partition coefficients"],
+ y = pc_cov["Molecular weight"],
+ hover_name = pc_cov["Compound name"],
+ color = pc_cov["Part_coef_group"],
+ width =800,
+ height =400,
+ title ="Partition coefficients vs. molecular weights for compounds used in COVID-19 clinical trials")
+
+fig.update_layout(
+ title =dict(
+ font =dict(
+ size =15)),
+ title_x =0.5,
+ margin =dict(
+ l =20, r =20, t =40, b =3),
+ xaxis =dict(
+ tickfont =dict(size =9),
+ title ="Partition coefficients"
+ ),
+ yaxis =dict(
+ tickfont =dict(size =9),
+ title ="Molecular weights"
+ ),
+ legend =dict(
+ font =dict(
+ size =9)))
+
+fig.show()
+
+
+
+
+
+
+
+
+
Molecular weights vs. Complexity
+
+
fig = px.scatter(x = pc_cov["Molecular weight"],
+ y = pc_cov["Complexity"],
+ hover_name = pc_cov["Compound name"],
+#color = pc_cov["Part_coef_group"],
+ width =800,
+ height =400,
+ title ="Molecular weights vs. complexity for compounds used in COVID-19 clinical trials")
+
+fig.update_layout(
+ title =dict(
+ font =dict(
+ size =15)),
+ title_x =0.5,
+ margin =dict(
+ l =20, r =20, t =40, b =3),
+ xaxis =dict(
+ tickfont =dict(size =9),
+ title ="Molecular weights"
+ ),
+ yaxis =dict(
+ tickfont =dict(size =9),
+ title ="Complexity"
+ ),
+ legend =dict(
+ font =dict(
+ size =9)))
+
+fig.show()
+
+
+
+
+
+
+
+
+
+
Export prepared dataset
+
Two of the possible options to export the dataset for use in a Shiny app could be:
+
+
Convert Polars dataframe into a Pandas dataframe, so that it could be imported into the app for use (Polars not directly supported in Shiny for Python yet, but we could use its to_pandas() function to coerce an object e.g. a dataframe to be converted into a Pandas dataframe).
+
Another option was to save Polars dataframe as .csv file, then read in this file in the app.py script by using Pandas (which was the method I used for this particular app)
+
+
```{python}
+# --If preferring to use Pandas--
+# Convert Polars df into a Pandas df if needed
+df_name = df_name.to_pandas()
+
+# Convert the Pandas df into a csv file using Pandas
+df_name.to_csv("csv_file_name.csv", sep =",")
+
+# --If preferring to use Polars--
+# Simply write a Polars dataframe into a .csv file
+df_name.write_csv("csv_file_name.csv", separator =",")
+```
+
+
+
+
+ ]]>
+ Python
+ Shiny
+ Polars
+ Plotly
+ PubChem
+ Cheminformatics
+ https://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_data_prep.html
+ Sun, 07 May 2023 12:00:00 GMT
+
+
+ Shiny app in R
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/12_Shiny_app_chembl/ShinyAppChembl.html
+
+
Back story
+
It has been a long while since I’ve used R in my projects. Last year during the time when I bravely embraced the role of rotating curator for @WeAreRLadies on Twitter around end of October, I did mention that I wanted to learn Shiny. I haven’t forgotten about this actually. So as promised, here it is, my first Shiny app in R, which is really a very simple interactive web app about small molecules from ChEMBL database. The URL to reach this app, ShinyAppChembl, is at: https://jhylin.shinyapps.io/ShinyAppChembl/. It shows a selected set of physicochemical properties for the curated small molecules in different max phases in boxplot formats. Note: it may take a few minutes to load the plot when first opening the app.
+
+
+
+
The process
+
Since I haven’t been using a lot of R lately, I just wanted to document how I approached this Shiny app framework, as part of my self-directed learning for R that started around mid-2022. The first place I went to was not Google’s Bard or OpenAI’s ChatGPT, as I was trying to preempt a scenario where if both of these options were temporarily down, what would I do to learn a new tool. So I visited the Shiny website first, and literally started from the “Get Started” section there, then tried to read through the lessons provided. I gathered a quick overview about the core components within a Shiny app, which were the user interface, server logic and the call to run or create app in the end, and thought to get started from there.
+
One of the most helpful online books called, “Mastering Shiny” had clarified a few coding issues for me. The reactivity section in the book was very useful as well to help with understanding the interactivity concept in the app. The best and also the hardest thing at this stage after reading some of the information was to actually start coding for the app in RStudio IDE, which I did soon after.
+
+
+
+
Trials-and-errors
+
Initially, I’ve noticed in the gallery section from the Shiny website that some of the basic Shiny apps had plots produced with R code using S3 method - the type with class ‘formula’, such as boxplot(formula e.g. y ~ group, data, and so on). So I started with this first and ended up with a draft version shown below:
+
+
+
+First draft app using S3 method in R code to plot boxplots - screenshot taken by author
+
+
+
+
I then tried the ggplot2 version, which I preferred to use. However, I kept on hitting a roadblock repeatedly (as shown in the image below):
+
+
+
+Second draft app using ggplot2 boxplot format - screenshot taken by author
+
+
+
I ended up working through this issue of not being able to display the boxplots properly over at least two days, where I tried to figure out how to change the code so that the boxplots would appear as the output in the app. I actually wrote a plot function code (as shown below) before working on the app.R file, in order to trial plotting the boxplots, making sure that the code worked before using it in the app.R file.
+
```{r}
+dfBoxplot <-function(var) {
+ label <- rlang::englue("{{var}} vs. Max Phases of small molecules")
+
+ chembl %>%
+select(`Max Phase`, {{ var }}) %>%
+ggplot(aes(x =`Max Phase`, y = {{ var }})) +
+geom_boxplot(aes(group =cut_width(`Max Phase`, 0.25),
+colour =`Max Phase`),
+outlier.alpha =0.2) +
+labs(title = label)
+}
+```
+
Once I made sure this code worked, I transplanted the code into the server section of the app.R file, however it wasn’t that simple obviously. Through the process of more trials-and-errors, I managed to figure out the code for the plot output in the final version, which was not the same as the function code above, but more like this.
+
```{r}
+ output$BPlot <-renderPlot({
+
+ggplot(chembl, aes(`Max Phase`, .data[[input$variable]])) +
+geom_boxplot(aes(group =cut_width(`Max Phase`, 0.25),
+colour =`Max Phase`), outlier.alpha =0.2) +
+labs(title ="Distributions of physicochemical properties against max phases",
+caption ="(based on ChEMBL database version 31)") +
+theme_minimal()
+
+ }, res =96) %>%bindCache(chembl$`Max Phase`, input$variable)
+```
+
I then read about the section on “Tidy evaluation” in the “Mastering Shiny” book, which had thoroughly described the problems I’ve encountered (and which I wished I had actually read this section before and not after hitting the roadblock…). So I’d highly recommend new users to read this section and also the rest of the book if Shiny’s also new to you.
+
+
+
+
Final app
+
The final app now looks like this:
+
+
+
+Screenshot taken by author
+
+
+
+
+
+
App deployment
+
After I got the app working, I looked into where I could deploy the app, since my main goal was to learn and share my work. At first, I went to the Shiny section on the Quarto website to see if it was possible to deploy the app in Quarto. However, after reading through several questions and answers in relation to Shiny apps and Quarto website, it was obvious that it was still not possible yet to deploy the app in an interactive way on Quarto websites (but it was mentioned in Posit community that this was being looked into, so I’m looking forward to the day when we can do exactly that in the future). This means that currently, there will only be an app image showing up in a Quarto document at most. I ended up choosing shinyapp.io to deploy my first Shiny app for now.
+
+
+
+
About the boxplots
+
Since the main goal of this post is more on the process of producing a simple Shiny app for a new comer, I won’t go into the fine details to describe how these boxplots differ between different max phases. Also as a side note, I’m aware that some experts in data visualisations might not really like boxplots in general, but for my case, I’ve got molecules in different max phases where a boxplot is presented for each max phase lining up next to each other. Therefore, in a way, some relative comparisons or differences could be drawn visually in the first glance, although other graph types such as density plots or heat maps might be better options.
+
I’ll focus on the “QED Weighted” variable here, as it’s a physicochemical property that has combined several molecular features together as a score (please refer to this post - section: “Some exploratory data analysis” for details about this QED weighted score). For all the boxplots shown when “QED Weighted” is selected from the drop-down box, max phase 4 molecules obviously have higher QED weighted scores in general than all of the other max phases. This is especially clear when comparing the medians between them, with max phase 4 small molecules having a median QED weighted score of more than 0.5, and the rest of the other max phases had 0.5 or below. The higher the QED weighted scores, the more druglike the molecules will be, and for max phase 4 molecules, they are mostly prescription medicines that have already reached approval and are already being widely prescribed. So this makes sense as this is being reflected in the boxplots for these ChEMBL small molecules.
+
+
+
+
Final words
+
Finally, I’m quite pleasantly surprised that there is also a Shiny in Python version, which has a Shinylive option to deploy Shiny app interactively in GitHub Gist and so on… I’ll most likely need to read further into this and make this as my next project. This is also a nice break from my recent machine learning projects, which I’ll try to return to once I’ve had enough fun with Shiny!
+
Thanks for reading.
+
+
+
+
+ ]]>
+ R
+ Shiny
+ ChEMBL database
+ Cheminformatics
+ https://jhylin.github.io/Data_in_life_blog/posts/12_Shiny_app_chembl/ShinyAppChembl.html
+ Thu, 06 Apr 2023 12:00:00 GMT
+
+
+
+ Pills dataset - Part 3
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_evcxr_polars_plotly_final.html
+
+
Background
+
The aim of this final part (part 3) for the pills dataset was really for me to start using Rust in a beginner-friendly way. Overall, this trilogy (parts 1 - 3) for the pills dataset formulated an overview of how to use Polars in Python (mainly), Pandas in Python (smaller section) and Polars in Rust (even little less as this was new to me) with Plotly. Over time, I’ve been finding myself learning more optimally by doing and applying, rather than just reading and thinking, so I’ve got myself started in this very new programming language, Rust, to get some familiarities. I anticipated that I would still work with Python and R mainly in the near future, so that I’m not diverting too much and would be at least proficient in at least one programming language.
+
My very initial idea was to integrate Rust-Polars, Plotly in Rust (Plotly.rs) and Jupyter-Evcxr together, and see if I could get a simple data visualisation out of a small dataset. Although the idea sounded simple enough, I was actually quite stuck at the step of importing one of the columns as x-axis variables in Rust-Polars to Plotly.rs. I figured it might possibly be due to my very lack-of-knowledge and lack-of-familiarities with Rust (I do need to continue reading the Rust programming language book), Polars (I’m better with Python-Polars actually), Plotly.rs and also Evcxr. Another possibility could be that Plotly.rs mainly had ndarray support, and Polars was not mentioned explicitly in Plotly.rs so my guess was that these two might not flow very well together. Also, Polars itself was constantly evolving and growing as well.
+
So I’ve decided to leave things as how it would be for now, before I delayed this post any further. If I happened to figure out how to do this in the future, then I’ll come back to update this last part of the project. While I was tackling this little issue mentioned above, somehow I’ve managed to deconstruct Polars dataframe in Rust in Evcxr. So I’ll show a little bit about it below. One slightly good news that came out from all of this, was that I’ve managed to import the other column as y-axis variables, which contained numbers, without problems. I’ve also figured out the Rust code to convert Series/ChunkedArray to vectors in Rust IDEs (e.g. VS Code, and quite a few others). So I did learn a few things while completing this post, and hoped I could expand further on this later.
+
Note: I’ve published all Rust code as print-only in Quarto markdown file, since it’s not possible to run them in RStudio IDE (Rust was not supported). So all Rust code were originally run on Jupyter Lab in MacOS, with code outputs being captured as screenshots, which were shown as photos in this post. Here’s the link to the .ipynb file in the GitHub repository for this portfolio website (or alternatively, you could access it from the GitHub icon link at the top of the web page), in case anyone wanted to see the full .ipynb version.
+
+
+
+
Import dependencies
+
These dependencies were known as crates in the world of Rust. I’d also like to think of them as libraries or packages we would install or import in Python and R. So this step was necessary before I even started anything decent in Rust. Similar things would also apply to Rust IDEs as well since I’ve played a little bit in VS Code previously.
+
```{rust}
+// Set up required dependencies
+:dep ndarray ="0.15.6"
+```
+
```{rust}
+:dep plotly = { version =">=0.8.0", features = ["plotly_ndarray"]}
+```
+
```{rust}
+// May take a few minutes to load polars crate (might depend on your machine specs)
+:dep polars = { version =">=0.26.0", features = ["lazy", "csv-file", "strings", "dtype-duration", "dtype-categorical", "concat_str", "rank", "lazy_regex", "ndarray"]}
+```
I’ve tested plotting in Plotly.rs after a few trials and errors at the beginning, but luckily I’ve spotted the ndarray support from the Plotly.rs book soon enough to figure out that I could convert the “count” column into a ndarray first, which was shown in the code below.
+
```{rust}
+// Switch Polars dataframe into 2D array
+// Ensure "ndarray" was added as one of the features for polars under dependencies
+
+/*Example from Polars documentation:
+let df = DataFrame::new(vec![a, b]).unwrap();
+let ndarray = df.to_ndarray::<Float64Type>().unwrap();
+println!("{:?}", ndarray);
+*/
+
+//Note: ndarray for numbers only, not strings, so only "count" column was converted
+let ndarray = df.to_ndarray::<Float64Type>().unwrap();
+println!("{:?}", ndarray);
+```
+
+
+
+Photo by author
+
+
+
+
+
+
Deconstructing Polars dataframe in Rust
+
Because “to_ndarray” was only for numerics and not strings, I ran into a problem trying to figure out how to best import this other “Colour” column into Plotly.rs. This led to my little convoluted journey to work with Polars dataframe in Rust, trying to see if I could convert the “Colour” column into a vector (which might not be the best way to do it, but as part of my Rust learning, I went for it anyway). I’ve subsequently tried plotting the “count” column in ndarray as a vector with success, based on the reference from Plotly.rs book that variables for x or y-axis could be placed into a vector by using a vector macro. Eventually, I didn’t quite achieve my goal but I’ve managed to break down or convert the Polars dataframe into different formats.
+
```{rust}
+// Select specific column or series by position
+let Colours = df[0].clone();
+
+//Alternative way to select specific column or series by name
+//let u =df.select_series(&["Colour"]);
+```
+
```{rust}
+Colours
+```
+
+
+
+Photo by author
+
+
+
There was a mention of storing series (column) in a vec (as series vector, not vector for strings) in Polars’ documentation, which I’ve tried to plot in Plotly.rs, but it unfortunately failed to work. One of my guesses could be due to the data type used for vector, as Rust was a very type-specific programming language, which also brought its well-known memory safety and other benefits in the long run. My immediate thought was that it probably needed to be a vector for strings, not series, which might make it work. Then I was searching on StackOverflow for similar questions and answers, then I found something related to what I wanted to do from Polars documentations as shown below.
+
```{rust}
+// Adapted from: https://docs.rs/polars/latest/polars/docs/eager/index.html#series
+// Extracting data:
+// To be able to extract data out of Series,
+// either by iterating over them or converting them to other datatypes like a Vec<T>,
+// we first need to downcast them to a ChunkedArray<T>.
+// This is needed because we don't know the data type that is held by the Series.
+
+/*use polars::prelude::*;
+ use polars::df;
+
+ fn extract_data() -> PolarsResult<()> {
+ let df = df! [ "a" => [None, Some(1.0f32), Some(2.0)], "str" => ["foo", "bar", "ham"]]?;
+
+// first extract ChunkedArray to get the inner type.
+
+ let ca = df.column("a")?.f32()?;
+
+// Then convert to vec
+
+ let _to_vec: Vec<Option<f32>> = Vec::from(ca);
+
+// We can also do this with iterators
+
+ let ca = df.column("str")?.utf8()?;
+ let _to_vec: Vec<Option<&str>> = ca.into_iter().collect();
+ let _to_vec_no_options: Vec<&str> = ca.into_no_null_iter().collect();
+
+ Ok(())
+
+}*/
+```
+
Initially, I trialled the iterator function first.
+
```{rust}
+// Print out items in column by applying an iterator to it
+println!("{}", &Colours.iter().format("\n"));
+```
+
+
+
+Photo by author
+
+
+
Then, it took me quite a long time to just downcast Series into ChunkedArray, but somehow I’ve managed to figure out the code myself below. One of the likely reasons was due to my choice of using Evcxr, which required Rust code in slightly different formats than the ones in Rust IDEs (although almost the same).
+
```{rust}
+// Somehow worked out how to convert series to chunkedarray by accident!
+println!("{:?}", Colours.utf8().unwrap());
+```
+
+
+
+Photo by author
+
+
+
Then I moved onto trying to figure out how to convert or place a ChunkedArray into a vector, with the closest answer shown below. However, bear in mind that these Rust code were for Rust IDEs, and not for Evcxr, so this added slightly more complexities to what I was trying to do (perhaps I should just stick with Rust IDEs in the future…).
+
```{rust}
+//Adpated from StackOverflow - How to get a Vec from polars Series or ChunkedArray?
+//You can collect the values into a Vec.
+
+/*use polars::prelude::*;
+
+fn main() -> Result<()> { let s = Series::new("a", 0..10i32);
+
+ let as_vec: Vec<Option<i32>>=s.i32()?.into_iter().collect();
+
+//if we are certain we don't have missing values
+ let as_vec: Vec<i32> = s.i32()?.into_no_null_iter().collect();
+ Ok(())
+
+}*/
+```
+
I also found another way to iterate the ChunkedArray.
I also found out, randomly, how to slice strings for Series in Polars. By changing the number of letters to slice through in Some(), the strings or words would vary length accordingly. Here, I’ve used “15” so it covered all the colours (note: the longest would be 12 characters for combination colours).
+
```{rust}
+// Another method to use if needing to slice strings
+let x =Colours.utf8().unwrap().str_slice(0, Some(15));
+x
+```
+
+
+
+Photo by author
+
+
+
Lastly, before I got too carried away, I just wanted to show the method from Polars documentations that this was the Polars’ way to select a specific column from Polars dataframe.
+
```{rust}
+let ca =df.clone().lazy().select([cols(["Colour"])]).collect()?;
+ca
+```
+
+
+
+Photo by author
+
+
+
+
+
+
Plotting Polars dataframe in Plotly.rs
+
For the x-axis, eventually, I reverted for manual input due to the issue mentioned in the background section. So the colours from the “Colour” column were stored in a vector set up manually, rather than coming directly from the dataframe. While searching for answers, I’ve also learnt several other tricks, although not really solving the problem, they might still be useful in the future. For the y-axis, the ndarray for the “count” column was converted into a vector first before being fed into the trace (graph module), and thankfully the plot worked nicely.
+
```{rust}
+// MANUAL method:
+// Use vec! macro to create new vectors to hold x variables (words as strings)
+// Manually input the colour names (as ndarray is only for numbers)
+let x = vec!["RED", "ORANGE;BROWN", "YELLOW;WHITE", "ORANGE", "WHITE", "BLUE"];
+
+// Plot using ndarray, which is supported by Plotly.rs
+// Polars likely not supported yet
+// Convert ndarray (holding counts as y variables) into vector
+let y =ndarray.column(1).to_vec();
+
+// Use trace as a graph module,
+// choose type of plots needed for x & y variables called
+// Graph options e.g. Scatter, Line or Bar
+let trace = Scatter::new(x, y);
+
+// Set plot variable as mutable and initiate a plot
+let mut plot = Plot::new();
+// Add trace (graph) into the plot variable
+plot.add_trace(trace);
+
+// Specify the specs for plot
+let layout = Layout::new()
+// Choose height of graph
+.height(500)
+// Name x-axis
+.x_axis(Axis::new().title(Title::new("Colours")))
+// Name y-axis
+.y_axis(Axis::new().title(Title::new("Count")))
+// Add title of graph
+.title(Title::new("Frequency of colours in acetaminophen (paracetamol) oral dosage forms"));
+
+// Set the layout of the plot
+plot.set_layout(layout);
+
+// Display the plot in Jupyter Lab format
+// For Jupyter Notebook, use:plot.notebook_display();
+plot.lab_display();
+format!("EVCXR_BEGIN_CONTENT application/vnd.plotly.v1+json\n{}\nEVCXR_END_CONTENT", plot.to_json())
+```
+
+
+
+Photo by author
+
+
+
+
+
+
Conclusion
+
This last part was the hardest for me to execute out of all 3 parts (it likely took me a good whole week to figure out deconstructing Polars dataframe and trying to work with vectors), as Rust was completely new to me. At one point I thought about jumping back to Python, but I persisted and although I didn’t quite solve the string importation issue, I was somehow happy that I was at least able to see how this programming language could be applied in Polars dataframe library. I also got a taste of using Rust in data visualisations. All I wanted to show was that there were a variety of data tools to use, and knowing your tools of trade would be the most critical when working on different data projects as certain tools would only work the best for certain tasks and scenarios. This warm-up lesson in Rust was quite interesting and I might continue either in VS Code or Evcxr depending on my next topic of interest.
Plotly.rs GitHub repository: https://github.com/igiagkiozis/plotly (link to the Plotly.rs book can be found in “Converting columns into ndarrays” section)
+
+
+
+
+ ]]>
+ Data analytics projects
+ Pills dataset series
+ Rust
+ Polars
+ Plotly
+ Evcxr
+ Jupyter
+ https://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_evcxr_polars_plotly_final.html
+ Mon, 13 Feb 2023 11:00:00 GMT
+
+
+
+ Pills dataset - Part 2
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_polars_pills_df.html
+
+
Quick overview
+
Part 2 of this project aimed to look at the pills data up close, particularly into the types of dosage forms, colours, shapes and inactive excipients used in oral medications. Plotly was used as the main data visualisation library, which was followed by some text cleaning for a particularly busy column in the dataset. This was then completed with a section in the end to generate a small dataframe, preparing for a simple data visualisation in Rust-Evcxr for the final part of this project (part 3).
One way to avoid switching Polars dataframe to a Pandas one, which could be one of the options to plot data from Polars dataframes in Plotly, was to call the x-axis and y-axis data directly from the dataframe as shown in the code below.
+
+
# scatter plot for colours, dosage forms & drug strengths
+fig = px.scatter(x = df_viz["Colour"],
+ y = df_viz["Dosage_form"],
+ color = df_viz["Colour"],
+ hover_name = df_viz["Drug_strength"],
+ width =900,
+ height =400,
+ title ="Oral dosage forms and colours of pills")
+
+# Update layout of the plot
+fig.update_layout(
+# Change title font size
+ title =dict(
+ font =dict(
+ size =15)),
+# Centre the title
+ title_x =0.5,
+# Edit margins
+ margin =dict(
+ l =20, r =20, t =40, b =3),
+# Change x-axis
+ xaxis =dict(
+ tickfont =dict(size =9),
+ title ="Colours"
+ ),
+# Change y-axis
+ yaxis =dict(
+ tickfont =dict(size =9),
+ title ="Dosage forms"
+ ),
+# Edit lengend font size
+ legend =dict(
+ font =dict(
+ size =9)))
+
+fig.show()
+
+
+
+
+
+
+
White was the most common colour, especially after zooming in the plot. Capsule was very commonly used as the oral dosage form of choice in this dataset.
+
+
+
+
Visualising shapes & colours in pills
+
+
fig = px.scatter(x = df_viz["Colour"],
+ y = df_viz["Shape"],
+ color = df_viz["Colour"],
+ hover_name = df_viz["Drug_strength"],
+ width =900,
+ height =400,
+ title ="Shapes and colours of pills")
+
+# Update layout of the plot
+fig.update_layout(
+# Change title font size
+ title =dict(
+ font =dict(
+ size =15)),
+# Centre the title
+ title_x =0.5,
+# Edit margins
+ margin =dict(
+ l =20, r =20, t =40, b =3),
+# Change x-axis
+ xaxis =dict(
+ tickfont =dict(size =9),
+ title ="Colours"
+ ),
+# Change y-axis
+ yaxis =dict(
+ tickfont =dict(size =9),
+ title ="Shapes"
+ ),
+# Edit lengend font size
+ legend =dict(
+ font =dict(
+ size =9)))
+
+fig.show()
+
+
+
+
+
+
+
Capsule was again the most common oral dosage shape used for pills in the dataset. Common colours included red, brown, blue, purple, pink, orange, green, white and yellow. Combination colours followed these common ones, which had a mixture of a variety of colours used simultaneously, likely to avoid confusions and errors in dispensings or administrations.
+
+
+
+
Visualising inactive excipients in pills
+
The messiest part of the data actually lied in the column of “Inactive_excipients”, with numerous different punctuations used inconsistently, such as forward slashes, commas and semi-colons. There were vast quantities of different inactive components used for oral dosage forms. Because of this, I had to spend a bit more time cleaning up the texts in order to find out what were the commonly used inactive ingredients in the end.
+
+
# Formulated a separate dataframe with just "Inactive_excipients"
+df_ie = df_new.select([pl.col("Inactive_excipients")])
+df_ie
+
+
+
+
+shape: (83925, 1)
+
+
+
+
+Inactive_excipients
+
+
+
+
+str
+
+
+
+
+
+
+"SILICON DIOXID...
+
+
+
+
+"SILICON DIOXID...
+
+
+
+
+"ANHYDROUS LACT...
+
+
+
+
+null
+
+
+
+
+"SORBITOL;ASPAR...
+
+
+
+
+"ANHYDROUS DIBA...
+
+
+
+
+"LACTOSE MONOHY...
+
+
+
+
+"FD&C BLUE NO. ...
+
+
+
+
+"silicon dioxid...
+
+
+
+
+"CROSPOVIDONE;M...
+
+
+
+
+"STARCH, CORN;C...
+
+
+
+
+"COPOVIDONE K25...
+
+
+
+
+...
+
+
+
+
+"SILICON DIOXID...
+
+
+
+
+"BUTYLATED HYDR...
+
+
+
+
+"MAGNESIUM CARB...
+
+
+
+
+"ACESULFAME POT...
+
+
+
+
+"CROSCARMELLOSE...
+
+
+
+
+"FD&C BLUE NO. ...
+
+
+
+
+"STARCH, CORN;H...
+
+
+
+
+"CARNAUBA WAX;F...
+
+
+
+
+"CITRIC ACID MO...
+
+
+
+
+"STARCH, CORN;D...
+
+
+
+
+"Cellulose, mic...
+
+
+
+
+"ANHYDROUS DIBA...
+
+
+
+
+
+
+
+
+
Text cleaning for inactive excipients column
+
To prepare this column for data visualisations, I used Polars’ string expressions (or more commonly known as regex - regular expressions) to try and tidy up the raw texts. When I did the text cleaning in Jupyter Lab initially, the line of code for .str.strip(” ,“) worked, but when I converted the .ipynb file into a .qmd (Quarto markdown) one, and used the same line, it failed to work due to the extra space in front of the comma. However, I got around the error by splitting it into two separate units as space and comma, and it worked without problem. One possible reason would be due to the reticulate package needed to run Python in RStudio IDE, and how Polars dataframe library was relatively newer than Pandas dataframe library, which meant certain features in Polars might not have been taken on board in the reticulate package (only my guess).
+
+
# Clean string texts
+# Convert uppercase letters into lowercase ones in the excipients column
+df_de = (df_ie.with_column(pl.col("Inactive_excipients").str.to_lowercase(
+# replace old punctuations (1st position) with new one (2nd position)
+ ).str.replace_all(
+";", ", "
+ ).str.replace_all(
+" /", ", "
+ ).str.replace_all(
+"/", ", "
+# Remove extra space & comma by stripping
+# In Jupyter notebook/lab - can combine space & comma: .str.strip(" ,")
+# For RStudio IDE - separate into two for this to work
+ ).str.strip(
+" "
+ ).str.strip(
+","
+# Split the texts by the specified punctuation e.g. comma with space
+ ).str.split(
+ by =", "
+# Create a new column with a new name
+ ).alias(
+"Inactive"
+ )
+# Explode the splitted texts into separate rows within the new column
+).explode(
+"Inactive"
+)
+)
+
+df_de
+
+
+
+
+shape: (840029, 2)
+
+
+
+
+Inactive_excipients
+
+
+Inactive
+
+
+
+
+str
+
+
+str
+
+
+
+
+
+
+"SILICON DIOXID...
+
+
+"silicon dioxid...
+
+
+
+
+"SILICON DIOXID...
+
+
+"edetate disodi...
+
+
+
+
+"SILICON DIOXID...
+
+
+"lactose monohy...
+
+
+
+
+"SILICON DIOXID...
+
+
+"magnesium stea...
+
+
+
+
+"SILICON DIOXID...
+
+
+"cellulose"
+
+
+
+
+"SILICON DIOXID...
+
+
+"microcrystalli...
+
+
+
+
+"SILICON DIOXID...
+
+
+"starch"
+
+
+
+
+"SILICON DIOXID...
+
+
+"corn"
+
+
+
+
+"SILICON DIOXID...
+
+
+"sodium lauryl ...
+
+
+
+
+"SILICON DIOXID...
+
+
+"fd&c blue no. ...
+
+
+
+
+"SILICON DIOXID...
+
+
+"fd&c red no. 4...
+
+
+
+
+"SILICON DIOXID...
+
+
+"gelatin"
+
+
+
+
+...
+
+
+...
+
+
+
+
+"Cellulose, mic...
+
+
+"shellac"
+
+
+
+
+"Cellulose, mic...
+
+
+"propylene glyc...
+
+
+
+
+"Cellulose, mic...
+
+
+"ammonia"
+
+
+
+
+"Cellulose, mic...
+
+
+"fd&c blue no. ...
+
+
+
+
+"ANHYDROUS DIBA...
+
+
+"anhydrous diba...
+
+
+
+
+"ANHYDROUS DIBA...
+
+
+"ferric oxide r...
+
+
+
+
+"ANHYDROUS DIBA...
+
+
+"hypromelloses"
+
+
+
+
+"ANHYDROUS DIBA...
+
+
+"polyethylene g...
+
+
+
+
+"ANHYDROUS DIBA...
+
+
+"magnesium stea...
+
+
+
+
+"ANHYDROUS DIBA...
+
+
+"titanium dioxi...
+
+
+
+
+"ANHYDROUS DIBA...
+
+
+"talc"
+
+
+
+
+"ANHYDROUS DIBA...
+
+
+"ferric oxide y...
+
+
+
+
+
+
+
+
+
# Quick look at the dataframe to see before and after text cleaning
+print(df_de.glimpse())
+
+
Rows: 840029
+Columns: 2
+$ Inactive_excipients <Utf8> SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;, SILICON DIOXIDE;EDETATE DISODIUM;LACTOSE MONOHYDRATE;MAGNESIUM STEARATE;CELLULOSE, MICROCRYSTALLINE;STARCH, CORN;SODIUM LAURYL SULFATE;FD&C BLUE NO. 1;FD&C RED NO. 40;GELATIN;TITANIUM DIOXIDE;BUTYL ALCOHOL;
+$ Inactive <Utf8> silicon dioxide, edetate disodium, lactose monohydrate, magnesium stearate, cellulose, microcrystalline, starch, corn, sodium lauryl sulfate, fd&c blue no. 1
-
The following step was optional, but might be useful later, the web content could be saved as a file as shown below.
-
-
# Create a file by passing the request content into write () method
-# and save the dosage form table as a file in binary format
-withopen("FDA_dosage_form", "wb") asfile:
-file.write(data.content)
+
As shown above, the “Inactive_excipients” column was the original column for excipients, where the second column named, “Inactive” was the new column shown after the punctuation tidy-ups, string strip and row text explosion. The excipients were broken down into individual terms, rather than in massively long strings which might not make sense to some readers.
+
+
# Re-organise the dataframe to choose the cleaned "Inactive" column
+df_final = df_de.select(["Inactive"])
+df_final
+
+
+
+
+shape: (840029, 1)
+
+
+
+
+Inactive
+
+
+
+
+str
+
+
+
+
+
+
+"silicon dioxid...
+
+
+
+
+"edetate disodi...
+
+
+
+
+"lactose monohy...
+
+
+
+
+"magnesium stea...
+
+
+
+
+"cellulose"
+
+
+
+
+"microcrystalli...
+
+
+
+
+"starch"
+
+
+
+
+"corn"
+
+
+
+
+"sodium lauryl ...
+
+
+
+
+"fd&c blue no. ...
+
+
+
+
+"fd&c red no. 4...
+
+
+
+
+"gelatin"
+
+
+
+
+...
+
+
+
+
+"shellac"
+
+
+
+
+"propylene glyc...
+
+
+
+
+"ammonia"
+
+
+
+
+"fd&c blue no. ...
+
+
+
+
+"anhydrous diba...
+
+
+
+
+"ferric oxide r...
+
+
+
+
+"hypromelloses"
+
+
+
+
+"polyethylene g...
+
+
+
+
+"magnesium stea...
+
+
+
+
+"titanium dioxi...
+
+
+
+
+"talc"
+
+
+
+
+"ferric oxide y...
+
+
+
+
+
+
+
+
+
# Remove all cells with null values
+df_final = df_final.drop_nulls()
+
+
+
# Group the data by different inactive excipients with counts shown
+df_final = df_final.groupby("Inactive").agg(pl.count())
+df_final.head()
+
+
+
+
+shape: (5, 2)
+
+
+
+
+Inactive
+
+
+count
+
+
+
+
+str
+
+
+u32
+
+
+
+
+
+
+"low-substitute...
+
+
+4
+
+
+
+
+"sodium starch ...
+
+
+118
+
+
+
+
+" glyceryl dibe...
+
+
+3
+
+
+
+
+"aluminum chlor...
+
+
+27
+
+
+
+
+"mentha piperit...
+
+
+7
+
+
+
+
+
+
+
+
+
+
Inactive excipient counts
+
+
# Count each excipient and cast the whole column into integers
+df_final = df_final.with_column((pl.col("count")).cast(pl.Int64, strict =False))
+df_final
+
+
+
+
+shape: (1674, 2)
+
+
+
+
+Inactive
+
+
+count
+
+
+
+
+str
+
+
+i64
+
+
+
+
+
+
+"low-substitute...
+
+
+4
+
+
+
+
+"sodium starch ...
+
+
+118
+
+
+
+
+" glyceryl dibe...
+
+
+3
+
+
+
+
+"aluminum chlor...
+
+
+27
+
+
+
+
+"mentha piperit...
+
+
+7
+
+
+
+
+"epimedium gran...
+
+
+1
+
+
+
+
+" ethyl acetate...
+
+
+2
+
+
+
+
+"rutin"
+
+
+1
+
+
+
+
+"methacrylic ac...
+
+
+2106
+
+
+
+
+" calcium phosp...
+
+
+12
+
+
+
+
+"carbomer homop...
+
+
+28
+
+
+
+
+" tocopherol"
+
+
+2
+
+
+
+
+...
+
+
+...
+
+
+
+
+"methylcellulos...
+
+
+62
+
+
+
+
+"carbomer homop...
+
+
+27
+
+
+
+
+" red ferric ox...
+
+
+3
+
+
+
+
+"anhydrous lact...
+
+
+4
+
+
+
+
+"sorbic acid"
+
+
+195
+
+
+
+
+"ilex pedunculo...
+
+
+2
+
+
+
+
+" aminobenzoic ...
+
+
+1
+
+
+
+
+"polyvinyl alco...
+
+
+55
+
+
+
+
+"3-hexenyl acet...
+
+
+4
+
+
+
+
+"methacrylic ac...
+
+
+2
+
+
+
+
+"dihydroxyalumi...
+
+
+2
+
+
+
+
+"hydroxypropyl ...
+
+
+46
+
+
+
+
+
+
+
+
+
+
Overview of inactive excipients used in oral dosage forms
+
+
fig = px.scatter(x = df_final["Inactive"],
+ y = df_final["count"],
+ hover_name = df_final["Inactive"],
+ title ="Inactive excipients and their respective counts in pills")
+
+fig.update_layout(
+ title =dict(
+ font =dict(
+ size =15)),
+ title_x =0.5,
+ margin =dict(
+ l =20, r =20, t =40, b =10),
+ xaxis =dict(
+ tickfont =dict(size =9),
+ title ="Inactive excipients"
+ ),
+ yaxis =dict(
+ tickfont =dict(size =9),
+ title ="Counts"
+ ),
+ legend =dict(
+ font =dict(
+ size =9)))
+
+
+fig.show()
+
+
+
+
+
+
+
+
Frequently used inactive excipients
+
+
# Re-order the excipients with counts in descending order
+# Filter out only the ones with counts over 10,000
+df_ex = df_final.sort("count", reverse =True).filter((pl.col("count")) >=10000)
+df_ex.head()
+
+
+
+
+shape: (5, 2)
+
+
+
+
+Inactive
+
+
+count
+
+
+
+
+str
+
+
+i64
+
+
+
+
+
+
+"magnesium stea...
+
+
+58908
+
+
+
+
+"titanium dioxi...
+
+
+43241
+
+
+
+
+"unspecified"
+
+
+35744
+
+
+
+
+"silicon dioxid...
+
+
+34037
+
+
+
+
+"starch"
+
+
+32501
+
+
+
+
+
+
+
+
+
fig = px.bar(x = df_ex["Inactive"],
+ y = df_ex["count"],
+ color = df_ex["Inactive"],
+ title ="Commonly used inactive excipients in pills")
+
+fig.update_layout(
+ title =dict(
+ font =dict(
+ size =15)),
+ title_x =0.5,
+ margin =dict(
+ l =10, r =10, t =40, b =5),
+ xaxis =dict(
+ tickfont =dict(size =9),
+ title ="Inactive excipients"
+ ),
+ yaxis =dict(
+ tickfont =dict(size =9),
+ title ="Counts"
+ ),
+ legend =dict(
+ font =dict(
+ size =9)))
+
+fig.show()
+
+
+
+
-
-
-
-
Transform web-scraped data into dataframe
+
The text cleaning might not be perfect at this stage, but I think I’ve managed to get most of the core texts cleaned into a more sensible and readable formats. From what I’ve worked out here, the most frequently used inactive ingredient was magnesium stearate, which was followed by titanium dioxide, and then interestingly “unspecified”, which was exactly how it was documented in the original pillbox dataset at the beginning. I didn’t go further digging into what this “unspecified” inactive excipients might be, as in whether it meant it in a singular or plural forms. So this still remained a mystery at this stage, but if all these oral medications were FDA-approved, we would’ve hoped each and everyone of these pills would be verified in safety, quality and effectiveness before they entered into the market for wide prescriptions. In the worst case, each therapeutic drug should also have post-marketing surveillance, for long-term safety monitoring.
-
-
Using Pandas dataframe library
-
-
Pandas.append()
-
The original pandas.append() method was going to be deprecated in future versions of Pandas. This old method was shown as below:
-
```{python}
-# Create an empty dataframe with columns named "Dosage_form" & "Code"
-dosage_form = pd.DataFrame(columns = ["Dosage_form", "Code"])
-
-# Create a loop to find all <tr> tags in the soup object (scraped html content)
-for row in soup.find_all("tr"):
-# Set the columns to contain contents under <td> tags by searching all rows
- col = row.find_all("td")
-# if columns are not an empty list,
-# add the texts under columns in specified orders
-if (col != []):
- dosage = col[0].text
- code = col[1].text
-
-# Append each text item into the dosage_form dataframe
-dosage_form = dosage_form.append({"Dosage_form":dosage, "Code":code}, ignore_index =True)
-
-# Show dataframe
-dosage_form
-```
-
This method might still work currently, however, the newer and recommended methods would be to use the pandas.concat() method as shown below.
-
-
Pandas.concat()
-
First example:
-
-
# Create an empty dictionary
-dict= []
-
-# Create a loop to iterate through html tags from the soup (scraped html content)
-# find all html tags that began with <tr>
-for row in soup.find_all("tr"):
-# each column would hold the items under <td> tags
- col = row.find_all("td")
-if (col != []):
-# dosage form in column 1
- dosage = col[0].text
-# code in column 2
- code = col[1].text
-# Append each dosage form & code into the dictionary
-dict.append({"DosageForm": dosage, "dosage_form": code})
-
-# Check if the loop was iterating through the html tags
-# and that it was appending each dosage form & code into the dictionary
-# Uncomment line below
-#print(dict)
-
-# Create an empty dataframe with the column names wanted
-dosage_form = pd.DataFrame(columns = ["DosageForm", "dosage_form"])
-
-# Concatenate the dosage_form dataframe with the dataframe converted from dict
-df_new = pd.concat([dosage_form, pd.DataFrame.from_dict(dict)])
-
-# Print the combined dataframe df_new
-df_new
-
+
+
+
Create a small dataframe for data visualisation in Rust-Evcxr
+
All acetaminophens were filtered out in the “Drug_strength” column and all duplicates were removed in the dataset.
I’ve opted for finding out the different types of colours with their respective counts in oral acetaminophen, or also known as paracetamol in some other countries.
Second example by using pd.from_dict() method, which might have less lines of code:
-
-
# Create an empty dictionary
-dict= []
-
-# Create a loop to iterate through html tags from the soup (scraped html content)
-# find all html tags that began with <tr>
-for row in soup.find_all("tr"):
-# each column would hold the items under <td> tags
- col = row.find_all("td")
-if (col != []):
-# dosage form in column 1
- dosage = col[0].text
-# code in column 2
- code = col[1].text
-# Append each dosage form & code into the dict
-dict.append({"DosageForm": dosage, "dosage_form": code})
-
-# Check if the loop was working to iterate through the html tags
-# and that it was appending each dosage form & code into the dictionary
-# Uncomment line below
-#print(dict)
-
-# Convert the dictionary into a dataframe
-df_new = pd.DataFrame.from_dict(dict)
-
-# Print the dataframe df_new
-df_new
-
+
+
fig = px.scatter(x = df_ac["Colour"],
+ y = df_ac["count"],
+ size = df_ac["count"],
+ color = df_ac["Colour"],
+ title ="Frequency of colours in acetaminophen (paracetamol) oral dosage forms"
+ )
+
+fig.update_layout(
+ xaxis =dict(
+ title ="Colours"
+ ),
+ yaxis =dict(
+ title ="Counts"
+ )
+)
+
+fig.show()
+
-
+
+
+
I’ve decided to keep the dataframe very simple for part 3 as my original intention was to trial plotting a graph in Evcxr only (nothing fancy at this stage), and also to gain some familiarities with Rust as another new programming language for me. Readers might notice that I’ve opted for a scatter plot in Plotly (in Python3 kernel) here for this last dataframe, and when we finally got to part 3 (hopefully coming soon as I needed to figure how to publish Rust code in Quarto…), I might very likely revert this to a bar graph (in Rust kernel), due to some technical issues (slow dependency loading, and somehow with Plotly.rs in Evcxr, the scatter graph looked more like scatter line graph instead… more stories to follow) and being a new Rust-Evcxr user. At the very least, I’ve kind of tried something I’ve planned for, although not looking very elegant yet, with rooms for improvements in the future.
+
+
+
+
+ ]]>
+ Data analytics projects
+ Pills dataset series
+ Polars
+ Python
+ Plotly
+ Jupyter
+ https://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_polars_pills_df.html
+ Mon, 30 Jan 2023 11:00:00 GMT
+
+
+
+ Pills dataset - Part 1
+ Jennifer HY Lin
+ https://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_polars_pills_ws.html
+
+
Introduction
+
As mentioned in my last project, I’ve tried using Evcxr, which provided a way to use Rust interactively in a Jupyter environment. The name, “Evcxr”, was quite hard to remember at first. It was pronounced as “e-vic-ser” according to the author, which I’ve randomly come across in an online tech interview when I was looking into it. I’ve also sort of worked out a way to memorise its spelling by taking specific letters out of “evaluation context for rust” (which was what it was called in its GitHub repository).
+
For users of Jupyter Notebook/Lab and Python, they might be quite used to the working speed of the cell outputs. However, one thing I’ve noticed when I was using Evcxr or Rust kernel in Jupyter Lab was that the speed of cell outputs was noticeably slower (especially at the beginning while loading all the dependencies required). The speed improved when loading external crates and modules, and generally it was faster afterward.
+
Due to this reason (note: I did not look into any other optimising strategies for this and this could be restricted to my computer hardware specs, so this might differ for other users), I think Evcxr was not ideal for a very large and complex data science project yet (however if its ecosystem kept developing, it might be improved in the future). One thing of note was that when I was combing through issues in Evcxr’s GitHub repository, someone mentioned the slow compile time of the Rust compiler, which would have likely caused the snail speed, but knowing that the actual program running speed was blazingly fast, some sacrifice at the beginning made sense to me. Overall, Rust was really a systems programming language with memory efficiency (with no garbage collector), type safety and concurrency as some of its notable advantages.
+
Because of the dependency loading issue in the Jupyter environment, and also knowing there was already a dataframe library built from Rust, I’ve opted to use Polars-Python again for the data wrangling part of this project. This was also accompanied by the good old Pandas library as well (under the section of “Transform web-scraped data into dataframe” if anyone wants to jump to that part to see the code). I then went on to trial using Rust via Evcxr for data visualisation based on a small dataframe by using Plotly.rs. This project would be separated into 3 parts:
+
+
Part 1: Initial pillbox dataset loading and web-scraping
+
Part 2: Data wrangling and mining for data visualisations
+
Part 3: Using Rust for data visualisation
+
+
The main reason I wanted to try Evcxr was that I could see the potential of using Rust interactively to showcase the results in a relatively fast and efficient manner. This meant specific data exploratory results could reach wider audience, leading to more impacts in different fields, in a very broad term. Oppositely, for more specific users such as scientists or engineers, this meant experiments could be carried out in a safe and efficient manner, with test results readily available for future work planning.
+
+
+
Download dataset
+
This time the dataset was spotted from Data Is Plural, specifically the 2022.11.30 edition. The section I was interested in was the first paragraph at the top, about “Pills”. By going into one of the links provided in the paragraph, this brought me to the Pillbox dataset from the US National Library of Medicine (NLM). The .csv file was downloaded via the “Export” button at the top right of the webpage.
+
This pillbox dataset was actually retired since 28th January 2021, but was still available for educational or research purposes only. Therefore, it was not recommended for pill identifications as the dataset was not up-to-date. Alternative resources such as DailyMed would be more appropriate for readers in the US (as one of the examples). For readers in other countries, local health professionals and resources would be recommended for up-to-date information.
+
-
-
Using Polars dataframe library
-
Polars dataframe library also had a from_dict() method that could convert dictionary into a dataframe as shown below:
-
-
# Create an empty dictionary
-dict= []
-
-# Create a loop to iterate through html tags from the soup (scraped html content)
-# find all html tags that began with <tr>
-for row in soup.find_all("tr"):
-# each column would hold the items under <td> tags
- col = row.find_all("td")
-if (col != []):
-# dosage form in column 1
- dosage = col[0].text
-# code in column 2
- code = col[1].text
-# Append each dosage form & code into the dict
-dict.append({"DosageForm": dosage, "dosage_form": code})
-
-# Check if the loop was iterating through the html tags
-# and that it was also appending each dosage form & code into the dictionary
-# Uncomment line below
-#print(dict)
-
-# Convert dictionary to dataframe
-new_df = pl.from_dicts(dict)
-new_df
-
+
+
Importing library & dataset
+
+
# Install/upgrade polars if needed (uncomment the line below)
+#pip install --upgrade polars
+
+
+
import polars as pl
+
+
+
# Check version of polars (uncomment line below)
+#pl.show_versions()
When importing pillbox.csv file initially, an error message actually came up that showed, “…Could not parse ‘10.16’ as dtype Int64 at column 7…”. One way to get around this was to add “ignore_errors” to bypass this error first in order to load the dataset first. This error could be fixed when checking and converting data types for columns.
+
-
-
-
Preparation of dataframe for data visualisation
-
Once we have the scraped dataframe ready, we could combine it with our original dataframe from the .csv file (the idea was basically doing dataframe join). Then the dosage form code column could be removed to make it easier to read.
-
-
# Join the two dataframes together
-df_final = df_med.join(new_df, on ="dosage_form")
-# Drop the column dosage_form which had code of each dosage form
-df_final = df_final.drop("dosage_form")
-df_final
-
+
+
Initial data wrangling
+
The Pillbox dataset link from NLM provided a list of column information for users. To quickly see what were the columns in the dataset, we could use “df.glimpse()” to read column names, data types and the first 10 items in each column.
A relatively simple dataset would be extracted first for these pills data since I was an inexperienced user of Rust. Therefore, I’ve selected only certain columns for this purpose.
+
+
df_med = df.select([# shapes of medicines
+"splshape_text",
+# colours of medicines
+"splcolor_text",
+# strengths of medicines
+"spl_strength",
+# inactive ingredients/excipients in medicines
+"spl_inactive_ing",
+# dosage forms of medicines e.g. capsules or tablets etc.
+"dosage_form"]
+ )
+df_med
Some data wrangling and converting a csv file into a parquet file
-
A .csv file tends to be separated by delimiters e.g. commas, semicolons or tabs. To read it properly, we can add a delimiter term in the code to transform the dataframe into a more readable format.
-
Another thing being added below is to deal with null values early - by filling in “None” and “” values in the dataframe as “null” first. This will save some hassles later on (I’ve encountered this problem when trying to convert column data types so found out this may be the best way to resolve it).
Below is a series of data checks and cleaning that’ll reduce the original .csv file size (about 664.8 MB) into something more manageable. My goal is to get a parquet file under 104 MB which can then be uploaded to GitHub without using Git large file storage (this will be the last resort if this fails).
-
I’m checking the “Type” column first.
-
-
df.group_by("Type").len()
-
-
-
-shape: (11, 2)
Type
len
str
u32
"Unknown"
18015
null
369155
"Cell"
47
"Gene"
77
"Oligonucleotide"
170
…
…
"Antibody"
974
"Enzyme"
118
"Small molecule"
1920366
"Oligosaccharide"
92
"Unclassified"
4
-
-
-
The dataframe is further reduced in size by filtering the data for small molecules only, which are what I aim to look at.
There are 5485 entries with “NONE” as “Structure Type” which means they have unknown compound structures or not recorded in either compound_structures table or protein_therapeutics table. These entries will be removed from df_sm first.
-
Next, I’m filtering the df_sm dataset further by restricting the filters to only small molecules and excluding all “NONE” structure types.
# Check "NONE" entries are removed/filtered
-df_sm.group_by("Structure Type").len()
-
-
-
-shape: (3, 2)
Structure Type
len
str
u32
"SEQ"
1
"MOL"
1914876
"BOTH"
4
-
-
-
I’ve tried filtering out data using “Inorganic flag” previously, however it turns out to be not so suitable - it’ll rule out a lot of preclinical compounds with max phase 0 or max phase > 1 compounds with no calculated physicochemical properties, which means there may not be enough training data to build a machine learning model. So I’m opting for the “Targets” column here by ruling out the ones with zero targets.
I have tried two main different ways where one is using the write_parquet() by only adding file compression level parameter (the “without partition” way), and the other one using use_pyarrow & pyarrow_options to partition datasets. The changes in parquet file size are shown in the following two tables.
Parquet file size changes with data partitions (note: original .csv file size is 664.8 MB)
-
-
-
-
-
-
-
-
-
Compression level
-
Data restrictions
-
File size
-
Number of entries
-
-
-
-
-
default
-
None
-
-
using “Max Phase” as partition column
-
max phase 0 > 104 MB
-
max phases 1-4: each < 104 MB
-
-
2,331,700
-
-
-
15
-
None
-
-
max phase 0 > 104 MB
-
max phase 1-4: each < 104 MB
-
-
2,331,700
-
-
-
20
-
None
-
-
similar sizes as mentioned above
-
-
2,331,700
-
-
-
default
-
None
-
-
using “Type” as partition column
-
“Small molecule” file size = 135.2 MB
-
-
2,331,700
-
-
-
-
Finally, it appears that the one with three data restrictions at compression level of 22 has produced a file at 100.4 MB. I’m reading this file below into a dataframe to see if it’s working.
So it looks like it does. The next series of posts will be about trying to use Polars dataframe library all the way with scikit-learn.
-
Note: the way I’ve compressed the original data file may not be the best as I’m losing some data along the way by restricting the number of data entries. There are definitely other better ways out there, please use this example with care.
-
- ]]>
- Machine learning projects
- Polars
- Python
- Jupyter
- ChEMBL database
- Cheminformatics
- https://jhylin.github.io/Data_in_life_blog/posts/21_ML1-1_Small_mols_in_chembl_update/ML1-1-1_chembl_cpds_parquet_new.html
- Tue, 03 Jan 2023 11:00:00 GMT
-
-
-
- Small molecules in ChEMBL database
- Jennifer HY Lin
- https://jhylin.github.io/Data_in_life_blog/posts/21_ML1-1_Small_mols_in_chembl_update/ML1-1-3_chembl_cpds_ml_model.html
-
+
Web scraping
+
This was not planned initially but this might make my life a lot easier if I could scrape the dosage form table found through the Pillbox link, since the dosage form column was full of C-letter code. These dosage form code were hard to understand, so once I’ve got the code along with corresponding dosage forms in texts, the web-scraped information would be converted into a dataframe for further data manipulations.
+
+
# Uncomment lines below to install libraries needed for web-scraping
+#!pip install requests
+#!pip install beautifulsoup4
+
Import libraries
-
This is the third post that follows on from the previous two about parquet file and data preprocessing, and it will need the following libraries to build and train a logistic regression (LR) model before using it to predict max phase outcome on a testing dataset by using scikit-learn.
-
-
## using magic pip to install sklearn & altair (somehow venv keeps switching off in vscode...)
-# %pip install -U scikit-learn
-# %pip install altair
-
-import sklearn
-print(f"scikit-learn version used is: {sklearn.__version__}")
-from sklearn import preprocessing, set_config
-from sklearn.model_selection import train_test_split
-from sklearn.linear_model import LogisticRegression
-from sklearn.pipeline import Pipeline
-from sklearn.preprocessing import StandardScaler
-import polars as pl
-print(f"polars version used is: {pl.__version__}")
-import altair as alt
-print(f"altair version used is: {alt.__version__}")
-import pickle
-import numpy as np
-
-
scikit-learn version used is: 1.5.0
+
+
import requests
+from bs4 import BeautifulSoup
+import pandas as pd
-
-
polars version used is: 1.9.0
+
I’ve opted for using Beautiful Soup as the web-scraping library in Python, along with the requests library to be able to make a URL request call to retrieve web information. There were of course many other tools available as well. A caveat to be taken into consideration was that when web-scraping, it was always recommended to check whether the information being scraped were under a specific copyright license and so on. In this case, I’ve checked that the dosage form table link - https://www.fda.gov/industry/structured-product-labeling-resources/dosage-forms was from US FDA and it was stated that the information (both texts and graphs) were not copyrighted (unless otherwise stated, for this particular web page, there was nothing stated along those lines), but a link to this webpage should be provided so that readers could access most current information in the future.
+
+
+
Send web requests
+
+
# Specify URL address with information intended for web-scraping
+url ="https://www.fda.gov/industry/structured-product-labeling-resources/dosage-forms"
+# Request the web information via requests library & save under a data object
+data = requests.get(url)
-
-
altair version used is: 5.4.1
+
+
+
Parse web content
+
+
# Parse the web content from the URL link by using Beautiful Soup
+soup = BeautifulSoup(data.content, "html.parser")
+
+
+
Print web content
+
+
# Print out the scraped web information
+print(soup.prettify())
The same set of data saved in the previous post will be read here using polars dataframe library.
-
-
df = pl.read_csv("df_ml.csv")
-df
-
-
-
-shape: (5_670, 9)
Max_Phase
Polar Surface Area
HBA
HBD
#RO5 Violations
QED Weighted
CX LogP
CX LogD
Heavy Atoms
i64
f64
i64
i64
i64
f64
f64
f64
i64
0
66.81
4
1
0
0.47
3.94
3.94
32
0
62.55
3
1
0
0.93
3.38
3.38
25
0
73.86
5
1
2
0.12
9.34
9.34
40
0
84.22
4
2
0
0.76
2.01
-0.19
26
0
40.46
4
0
0
0.62
4.0
4.0
26
…
…
…
…
…
…
…
…
…
1
128.03
8
2
0
0.49
2.09
1.86
34
1
0.0
0
0
0
0.0
0.0
0.0
0
1
74.02
6
1
0
0.68
3.65
2.3
30
1
94.83
4
3
0
0.44
1.2
-1.18
12
1
95.92
6
1
0
0.9
1.66
1.66
18
+
The following step was optional, but might be useful later, the web content could be saved as a file as shown below.
+
+
# Create a file by passing the request content into write () method
+# and save the dosage form table as a file in binary format
+withopen("FDA_dosage_form", "wb") asfile:
+file.write(data.content)
-
-
Logistic regression with scikit-learn
-
LR is one of the supervised methods in the statistical machine learning (ML) area. As the term “supervised” suggests, this type of ML is purely data-driven to allow computers to learn patterns from the input data with known outcomes in order to predict the same target outcomes for a different set of data that is previously unseen by the computer.
-
-
Define X and y variables
-
The dataset will be splitted into X (features) and y (target) variables first.
Note: no need to use to_numpy() as there’s a transform step included when using pipeline to create a LR model (also StandardScaler() going to be used). This also applies if using fit_transform() or transform() when not using pipeline - see scikit-learn reference on “transform”.
-
-
-
Prepare training and testing sets
-
Then the data will be further splitted into separate training and testing sets.
-
-
## Random number generator
-#rng = np.random.RandomState(0) - note: this may produce different result each time
-
-X_train, X_test, y_train, y_test = train_test_split(X, y, test_size =0.2, random_state =50)
-print('Training set:', X_train.shape, y_train.shape)
-print('Testing set:', X_test.shape, y_test.shape)
-
-
Training set: (4536, 8) (4536,)
-Testing set: (1134, 8) (1134,)
-
-
+
+
Transform web-scraped data into dataframe
+
+
Using Pandas dataframe library
+
+
Pandas.append()
+
The original pandas.append() method was going to be deprecated in future versions of Pandas. This old method was shown as below:
+
```{python}
+# Create an empty dataframe with columns named "Dosage_form" & "Code"
+dosage_form = pd.DataFrame(columns = ["Dosage_form", "Code"])
+
+# Create a loop to find all <tr> tags in the soup object (scraped html content)
+for row in soup.find_all("tr"):
+# Set the columns to contain contents under <td> tags by searching all rows
+ col = row.find_all("td")
+# if columns are not an empty list,
+# add the texts under columns in specified orders
+if (col != []):
+ dosage = col[0].text
+ code = col[1].text
+
+# Append each text item into the dosage_form dataframe
+dosage_form = dosage_form.append({"Dosage_form":dosage, "Code":code}, ignore_index =True)
+
+# Show dataframe
+dosage_form
+```
+
This method might still work currently, however, the newer and recommended methods would be to use the pandas.concat() method as shown below.
chaining preprocessing step with different transformers and estimators in one go where we only have to call fit and predict once on our data
-
avoiding data leakage from the testing set into the training set by making sure the same set of samples is used to train the transformers and predictors
-
avoiding missing out on the transform step (note: calling fit() on pipeline is equivalent to calling fit() on each estimator and transform() input data before the next step, plus StandardScaler() is going to be used in the pipeline as well - repeating myself here but this is just a gentle reminder…)
-
-
The example below uses Pipeline() to construct a pipeline that takes in a standard scaler to scale data and also a LR estimator, along with some parameters.
-
-
## Pipeline:
-
-# Ensure prediction output can be read in polars df
-set_config(transform_output="polars")
-
-# multi_class defaults to 'auto' which selects 'ovr' if the data is binary, or if solver='liblinear'
-# multi_class is deprecated in version 1.5 and will be removed in 1.7
-# this post uses sklearn version 1.5.0
-params_lr = {
-# solver for small dataset
-"solver": "liblinear",
-"random_state": 50
-}
-
-LR = Pipeline(steps=[
-# Preprocess/scale the dataset (transformer)
- ("StandardScaler", StandardScaler()), # can add set_output() if preferred
-# e.g. StandardScaler().set_output(transform="polars")
-# Create an instance of LR classifier (estimator)
- ("LogR", LogisticRegression(**params_lr))
- ])
-
-# can add set_output() if preferred e.g. LR.set_output(transform="polars")
-LR.fit(X_train, y_train)
-pred = LR.predict(X_test)
-LR.score(X_test, y_test)
-
-
0.689594356261023
+
+
Pandas.concat()
+
First example:
+
+
# Create an empty dictionary
+dict= []
+
+# Create a loop to iterate through html tags from the soup (scraped html content)
+# find all html tags that began with <tr>
+for row in soup.find_all("tr"):
+# each column would hold the items under <td> tags
+ col = row.find_all("td")
+if (col != []):
+# dosage form in column 1
+ dosage = col[0].text
+# code in column 2
+ code = col[1].text
+# Append each dosage form & code into the dictionary
+dict.append({"DosageForm": dosage, "dosage_form": code})
+
+# Check if the loop was iterating through the html tags
+# and that it was appending each dosage form & code into the dictionary
+# Uncomment line below
+#print(dict)
+
+# Create an empty dataframe with the column names wanted
+dosage_form = pd.DataFrame(columns = ["DosageForm", "dosage_form"])
+
+# Concatenate the dosage_form dataframe with the dataframe converted from dict
+df_new = pd.concat([dosage_form, pd.DataFrame.from_dict(dict)])
+
+# Print the combined dataframe df_new
+df_new
+
+
+
+
+
+
+
+
+
DosageForm
+
dosage_form
+
+
+
+
+
0
+
AEROSOL
+
C42887
+
+
+
1
+
AEROSOL, FOAM
+
C42888
+
+
+
2
+
AEROSOL, METERED
+
C42960
+
+
+
3
+
AEROSOL, POWDER
+
C42971
+
+
+
4
+
AEROSOL, SPRAY
+
C42889
+
+
+
...
+
...
+
...
+
+
+
153
+
TAMPON
+
C47892
+
+
+
154
+
TAPE
+
C47897
+
+
+
155
+
TINCTURE
+
C43000
+
+
+
156
+
TROCHE
+
C43001
+
+
+
157
+
WAFER
+
C43003
+
+
+
+
158 rows × 2 columns
-
During the pipeline building, I’ve figured out how to integrate set_output() in Polars, and noted that the best use case is to show the feature_names_in_ along with coef_ (scikit-learn reference). The first issue is that the feature names are being generated as “[x0, x1, x2…]”, which is not useful. One of the possible reasons could be because all the molecular features are not in strings (as they’re either i64 or f64), so the feature names are not shown - I’m actually unsure about this but this is just my guess.
-
One of the other ways I’ve tried is to use ColumnTransformer() within the pipeline (scikit-learn reference - code example folded below) but unfortunately it hasn’t worked as well as expected.
The pipeline above is the final version that works to show molecular feature names with their corresponding coefficients in a polars dataframe output. There are 3 options to add either set_config(transform_output="polars") or set_output(transform_output="polars") with the pipeline code - only really needing one line (and not all 3 - it’ll still work but probably unnecessary to add extra code). I’ve marked all 3 options in the pipeline code above.
-
-
-
Molecular features and coefficients
-
Next, I’m calling out the LR model used above in the pipeline as we want to get the feature names used for training and predicting along with their corresponding coefficients, and generate a bar chart to show their relationship (reference on plotting directly in Polars using Altair).
Once we have the scraped dataframe ready, we could combine it with our original dataframe from the .csv file (the idea was basically doing dataframe join). Then the dosage form code column could be removed to make it easier to read.
+
+
# Join the two dataframes together
+df_final = df_med.join(new_df, on ="dosage_form")
+# Drop the column dosage_form which had code of each dosage form
+df_final = df_final.drop("dosage_form")
+df_final
In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook. On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
# Save feature array as df
-lr_feat = pl.Series(log_reg.feature_names_in_).to_frame("Feature_names")
-# Explode df due to a list series - e.g. array([[1, 2, 3...]]) and not array([1, 2, 3...])
-lr_coef = pl.Series(log_reg.coef_).to_frame("Coef").explode("Coef")
-# Concatenate dfs horizontally
-df_feat = pl.concat([lr_feat, lr_coef], how="horizontal")
-
-# Using altair to plot feature names vs. coefficients
-df_feat.plot.bar(
- x="Coef",
-# -x = sorting in descending order, x = ascending
- y=alt.Y("Feature_names").sort("-x"),
-#color="Feature_names", #will create a legend if used
- tooltip="Coef",
-).configure_axis(
- labelFontSize=15,
- titleFontSize=15
-).configure_view(
- continuousWidth=600,
- discreteHeight=300
-)
#RO5 Violations, CXLogP, HBA and HBD all have positive weights or coefficients, when the rest of the molecular features (CXLogD, heavy atoms, polar surface area and QED Weighted) all have the negative coefficients. This is likely the equivalent of using the feature_importances_ in random forest I’m guessing. I’ve sorted the order of coefficients from highest to lowest in the chart.
-
Another way to get features names is from the pipeline as well but requires a step saving dataframe column names separately as an NumPy array first (scikit-learn reference). The previous way seems to save a bit more time on coding as there’s no need to do this, and also you can retrieve the coefficients of the features at the same time.
One way to get predicted probabilities of the samples in each outcome class (either 0 - not approved or 1 - approved) is via predict_proba() in scikit-learn.
Then we can convert the predicted probabilities into a polars dataframe, along with a statistics summary.
-
-
pl.DataFrame(y_mp_pre_proba).describe()
-
-
-
-shape: (9, 3)
statistic
column_0
column_1
str
f64
f64
"count"
1134.0
1134.0
"null_count"
0.0
0.0
"mean"
0.486442
0.513558
"std"
0.199652
0.199652
"min"
0.00459
0.044198
"25%"
0.341463
0.360326
"50%"
0.506803
0.493416
"75%"
0.639674
0.658537
"max"
0.955802
0.99541
-
-
-
Pickle LR pipeline
-
This last part is really for saving the LR pipeline for the next post on evaluating the LR model. I’ve talked a bit more about the security aspect of pickling files in this old post in case anyone’s interested.
-
-
# Pickle to save (serialise) the model in working directory (specify path if needed)
-pickle.dump(LR, open("LR.pkl", "wb")) # "wb" - write binary
-# Unpickle (de-serialise) the model
-LR2 = pickle.load(open("LR.pkl", "rb")) # "rb" - read binary
-# Use the unpickled model object to make prediction
-pred2 = LR2.predict(X_test)
-## Check unpickled model and original model are the same via Python's assertion method
-#assert np.sum(np.abs(pred2 - pred)) == 0
-## or alternatively use numpy's allclose()
-print(np.allclose(pred, pred2)) # note: pred = LR.predict(X_test) from original LR pipeline
-
-
True
-
+
Here, we could save the intended dataframe for data visualisation as a .csv file, so that further data wrangling and mining could be done later for part 2. This also avoided making request calls to the website again and again by extracting the scraped web information as a stand-alone file which could be imported when needed later on.
+
+
# Save the inital cleaned dataframe as .csv file
+# for use in a new .ipynb file with Rust kernel
+df_final.write_csv("pills.csv", sep =",")
Strength of evidence that the drug is metabolised by CYP3A4/5 (as quoted from above web link):
+
+
Strong Evidence: the enzyme is majorly responsible for drug metabolism.
+
Moderate Evidence: the enzyme plays a significant but not exclusive role in drug metabolism or the supporting literature is not extensive.
+
+
+
+
+
drug_class data sources
+
This information can be found in many national drug formularies, drug reference textbooks e.g. Martindale, American society of health-System pharmacists’ (ASHP) drug information (DI) monographs, PubChem, ChEMBL, FDA, Micromedex etc. or online drug resources such as Drugs.com. For the particular small dataset collected and used in the notebook, the following reference sources for ADRs also contain information on therapeutic drug classes too.
using the health professional version for ADRs and it usually contains ADR references from pharmaceutical manufacturers’ medicines information data sheets, ASHP DI monographs or journal paper references
+
+
2nd-line as separate data checks:
+
+
NZ formulary (nzf) - likely only available to NZ residents only; other national formularies should contain very similar drug information
drugs.com_uk_di - UK drug information section in Drugs.com (equivalent to pharmaceutical manufacturers’ medicines information data sheets)
+
+
two main types of occurrences/frequencies:
+
^^ - common > 10%,
+
^ - less common 1% to 10%,
+
(not going to include other ones with lower incidences e.g. less common at 0.1% to 1%, rare for less than 0.1% etc.)
+
+
+
+
+
Exceptions or notes for ADRs
+
+
nausea and vomiting applies to many drugs so won’t be included (almost every drug will have these ADRs, they can be alleviated with electrolytes replacements and anti-nausea meds or other non-med options; rash on the other hand can sometimes be serious and life-threatening e.g. Stevens-Johnson syndrome)
+
similar or overlapping adverse effects will be removed to keep only one adverse effect for the same drug e.g. adverse skin reactions, rash, urticaria - rash and urticaria will be removed as allergic skin reactions encompass both symptoms
+
for ADR terms with similar meanings, e.g. pyrexia/fever - fever is used instead (only one will be used)
+
ADR mentioned in common ADR category and repeated in the less common one will have the ADR recorded in the higher incidence rate (at > 10%) only
+
some ADRs can be dose-related or formulations-related e.g. injection site irritations or allergic reactions caused by excipients/fillers (aim is to investigate the relationships between ADRs and drugs via computational tools e.g. any patterns between ADRs & drugs so dose/formulations-related ADRS will not be recorded here)
+
some postmarketing adverse effects are for different age populations e.g. paediatric patients of up to 12 years of age or elderly people - for now all of them are labelled as “(pm)” to denote postmarketing reports and are not differentiated in age groups
+
+
+
Notes for specific drugs
+
+
hydrocortisone (a moderate CYP3A4 substrate), there are no reported ADR frequencies at all for its ADRs as they are entirely dependent on the dosage and duration of use (ADRs tend to be unnoticeable at appropriate low doses for short durations)
+
terfenadine (a strong CYP3A4 substrate) is actually withdrawn from the market in 1990s due to QT prolongations
+
lercanidipine (a moderate CYP3A4 substrate) has nil reported ADRs of more than 1% but has a few postmarketing reports recorded
+
telaprevir (a moderate CYP3A4 substrate) is usually administered within a combination therapy (e.g. along with peginterferon alfa and ribavirin)
+
quinine (a moderate CYP3A4 substrate) has all of its ADRs reported without frequencies. The most common ADRs are presented as a cluster of symptoms (known as cinchonism) and can occur during overdoses (usually very toxic) and also normal doses. These symptoms include “…tinnitus, hearing impairment, headache, nausea, vomiting, abdominal pain, diarrhoea, visual disturbances (including blindness), arrhythmias (which can have a very rapid onset), convulsions (which can be intractable), and rashes.” (as quoted from NZ formulary v150 - 01 Dec 2024)
+
ribociclib (a moderate CYP3A4 substrate) has a listed ADR of on-treatment deaths, which were found to be associated with patients also taking letrozole or fulvestrant at the same time and/or in patients with underlying malignancy
+
+
+
+
+
+
Abbreviations used
+
+
ws = withdrawal symptoms
+
ADH = antidiuretic hormone
+
pm = postmarketing reports
+
CNS = central nervous system
+
CFTR = cystic fibrosis transmembrane regulator
+
c_diff = Clostridioides/Clostridium difficile
+
ZE = Zollinger-Ellison
+
MTOR = mammalian target of rapamycin (protein kinase)
+
AST = aspartate transaminase/aminotransferase
+
ALT = alanine transaminase/aminotransferase
+
ALP = alkaline phosphatase
+
GGT = gamma-glutamyltransferase
+
RTI = respiratory tract infection
+
UTI = urinary tract infection
+
LDH = lactate dehydrogenase
+
dd = dose and duration-dependent
+
pm_HIV_pit = postmarketing reports for HIV protease inhibitor therapy
+
pm_hep_cyto = postmarketing reports in cancer patients where drug was taken with hepatotoxic/cytotoxic chemotherapy and antibiotics
+
+
+
+
+
+
+
+
+
+
+
+
+
+
\ No newline at end of file
diff --git a/posts/22_DL1_Simple_dnn_adrs/2_ADR_regressor.html b/posts/22_DL1_Simple_dnn_adrs/2_ADR_regressor.html
new file mode 100644
index 0000000..2a77ce9
--- /dev/null
+++ b/posts/22_DL1_Simple_dnn_adrs/2_ADR_regressor.html
@@ -0,0 +1,1132 @@
+
+
+
+
+
+
+
+
+
+Home - Building a simple deep learning model about adverse drug reactions
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
Building a simple deep learning model about adverse drug reactions
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
This notebook uses a venv created by using uv. Some of the code blocks have been folded to keep the post length a bit more manageable - click on the code links to see full code (this only applies to the HTML version, not the Jupyter notebook version).
+
+
+
Import libraries
+
+
+Code
+
import pandas as pd
+import torch
+import torch.nn as nn
+import torch.nn.functional as F
+from torch.nn.functional import one_hot
+from torch.utils.data import TensorDataset, DataLoader
+import numpy as np
+import datamol as dm
+import rdkit
+from rdkit import Chem
+from rdkit.Chem import rdFingerprintGenerator
+import useful_rdkit_utils as uru
+import sys
+from matplotlib import pyplot as plt
+print(f"Pandas version used: {pd.__version__}")
+print(f"PyTorch version used: {torch.__version__}")
+print(f"NumPy version used: {np.__version__}")
+print(f"RDKit version used: {rdkit.__version__}")
+print(f"Python version used: {sys.version}")
+
+
+
+
+
+
Import adverse drug reactions (ADRs) data
+
This is an extremely small set of data compiled manually (by me) via references stated in the dataframe. For details about what and how the data are collected, I’ve prepared a separate post as a data note (add post link) to explain key things about the data. It may not lead to a very significant result but it is done as an example of what an early or basic deep neural network (DNN) model may look like. Ideally there should be more training data features added or used.
+
+
data = pd.read_csv("All_CYP3A4_substrates")
+print(data.shape)
+data.head(3)
+
+
For drug with astericks marked in “notes” column, see data notes under “Exceptions for ADRs” section in 1_ADR_data.qmd (separate post).
+
I’m dropping some of the columns that are not going to be used later.
Before extracting data from ChEMBL, I’m getting a list of drug names in capital letters ready first which can be fed into chembl_downloader with my old cyp_drugs.py to retrieve the SMILES of these drugs.
+
+
+Code
+
string = df["generic_drug_name"].tolist()
+# Convert list of drugs into multiple strings of drug names
+drugs =f"'{"','".join(string)}'"
+# Convert from lower case to upper case
+for letter in drugs:
+if letter.islower():
+ drugs = drugs.replace(letter, letter.upper())
+print(drugs)
+
+
+
+
+Code
+
# Get SMILES for each drug (via copying-and-pasting the previous cell output - attempted various ways to feed the string
+# directly into cyp_drugs.py, current way seems to be the most straightforward one...)
+from cyp_drugs import chembl_drugs
+# Using ChEMBL version 34
+df_3a4 = chembl_drugs(
+'CARBAMAZEPINE','ELIGLUSTAT','FLIBANSERIN','IMATINIB','IBRUTINIB','NERATINIB','ESOMEPRAZOLE','OMEPRAZOLE','IVACAFTOR','NALOXEGOL','OXYCODONE','SIROLIMUS','TERFENADINE','DIAZEPAM','HYDROCORTISONE','LANSOPRAZOLE','PANTOPRAZOLE','LERCANIDIPINE','NALDEMEDINE','NELFINAVIR','TELAPREVIR','ONDANSETRON','QUININE','RIBOCICLIB','SUVOREXANT','TELITHROMYCIN','TEMSIROLIMUS',
+#file_name="All_cyp3a4_smiles"
+ )
+print(df_3a4.shape)
+df_3a4.head(3)
+
+## Note: latest ChEMBL version 35 (as from 1st Dec 2024) seems to be taking a long time to load (no output after ~7min),
+## both versions 33 & 34 are ok with outputs loading within a few secs
+
+
+
+
+
+
Merge dataframes
+
Next, I’m renaming the drug name column and merging the two dataframes together where one contains the ADRs and the other one contains the SMILES. I’m also making sure all drug names are in upper case for both dataframes so they can merge properly.
Then I’m parsing the canonical SMILES through my old script to generate these small molecules as RDKit molecules and standardised SMILES, making sure these SMILES are parsable.
+
+
+Code
+
# Using my previous code to preprocess small mols
+# disable rdkit messages
+dm.disable_rdkit_log()
+
+# The following function code were adapted from datamol.io
+def preprocess(row):
+
+"""
+ Function to preprocess, fix, standardise, sanitise compounds
+ and then generate various molecular representations based on these molecules.
+ Can be utilised as df.apply(preprocess, axis=1).
+
+ :param smiles_column: SMILES column name (needs to be names as "canonical_smiles")
+ derived from ChEMBL database (or any other sources) via an input dataframe
+ :param mol: RDKit molecules
+ :return: preprocessed RDKit molecules, standardised SMILES, SELFIES,
+ InChI and InChI keys added as separate columns in the dataframe
+ """
+
+# smiles_column = strings object
+ smiles_column ="canonical_smiles"
+# Convert each compound into a RDKit molecule in the smiles column
+ mol = dm.to_mol(row[smiles_column], ordered=True)
+# Fix common errors in the molecules
+ mol = dm.fix_mol(mol)
+# Sanitise the molecules
+ mol = dm.sanitize_mol(mol, sanifix=True, charge_neutral=False)
+# Standardise the molecules
+ mol = dm.standardize_mol(
+ mol,
+# Switch on to disconnect metal ions
+ disconnect_metals=True,
+ normalize=True,
+ reionize=True,
+# Switch on "uncharge" to neutralise charges
+ uncharge=True,
+# Taking care of stereochemistries of compounds
+# Note: this uses the older approach of "AssignStereochemistry()" from RDKit
+# https://github.com/datamol-io/datamol/blob/main/datamol/mol.py#L488
+ stereo=True,
+ )
+
+# Adding following rows of different molecular representations
+ row["rdkit_mol"] = dm.to_mol(mol)
+ row["standard_smiles"] = dm.standardize_smiles(str(dm.to_smiles(mol)))
+#row["selfies"] = dm.to_selfies(mol)
+#row["inchi"] = dm.to_inchi(mol)
+#row["inchikey"] = dm.to_inchikey(mol)
+return row
+
+df_p3a4 = df.apply(preprocess, axis =1)
+print(df_p3a4.shape)
+df_p3a4.head(3)
+
+
+
+
+
+
Split data
+
Random splits usually lead to overly optimistic models, where testing molecules are too similar to traininig molecules leading to many problems. This is further discussed in two other blog posts that I’ve found useful - post by Greg Landrum and post by Pat Walters.
+
Here I’m try out Pat’s useful_rdkit_utils’ GroupKFoldShuffle code (code originated from this thread) to split data (Butina clustering/splits). To do this, it requires SMILES to generate molecular fingerprints which will be used in the training and testing sets (potentially for future posts and in real-life cases, more things can be done with the SMILES or other molecular representations for machine learning, but to keep this post easy-to-read, I’ll stick with only generating the Morgan fingerprints for now).
+
+
+Code
+
# Generate numpy arrays containing the fingerprints
+df_p3a4['fp'] = df_p3a4.rdkit_mol.apply(rdFingerprintGenerator.GetMorganGenerator().GetCountFingerprintAsNumPy)
+
+# Get Butina cluster labels
+df_p3a4["butina_cluster"] = uru.get_butina_clusters(df_p3a4.standard_smiles)
+
+# Set up a GroupKFoldShuffle object
+group_kfold_shuffle = uru.GroupKFoldShuffle(n_splits=5, shuffle=True)
+
+# Using cross-validation/doing data split
+## X = np.stack(df_s3a4.fp), y = df.adverse_drug_reactions, group labels = df_s3a4.butina_cluster
+for train, test in group_kfold_shuffle.split(np.stack(df_p3a4.fp), df.adverse_drug_reactions, df_p3a4.butina_cluster):
+print(len(train),len(test))
+
+
+
+
+
+
Locate training and testing sets after data split
+
While trying to figure out how to locate training and testing sets after the data split, I’ve gone into a mini rabbit hole myself (a self-confusing session but gladly it clears up when my thought process goes further…). For example, some of the ways I’ve planned to try: create a dictionary as {index: butina label} first - butina cluster labels vs. index e.g. df_s3a4[“butina_cluster”], or maybe can directly convert from NumPy array to tensor - will need to locate drugs via indices first to specify training and testing sets, e.g. torch_train = torch.from_numpy(train) or torch_test = torch.from_numpy(test). It is actually simpler than this, which is to use pd.DataFrame.iloc() as shown below.
+
+
# Training set indices
+train
+
+
+
# What df_p3a4 now looks like after data split - with "fp" and "butina_cluster" columns added
+df_p3a4.head(1)
+
+
+
# Convert indices into list
+train_set = train.tolist()
+# Locate drugs and drug info via pd.DataFrame.iloc
+df_train = df_p3a4.iloc[train_set]
+print(df_train.shape)
+df_train.head(2)
Set up training and testing sets for X and y variables
+
This part involves converting X (features) and y (target) variables into either one-hot encodings or vector embeddings, since I’ll be dealing with categories/words/ADRs and not numbers, and also to split each X and y variables into training and testing sets. At the very beginning, I’ve thought about using scikit_learn’s train_test_split(), but then realised that I should not need to do this as it’s already been done in the previous step (obviously I’m confusing myself again…). Essentially, this step can be integrated with the one-hot encoding and vector embeddings part as shown below.
+
There are three coding issues that have triggered warning messages when I’m trying to figure out how to convert CYP strengths into one-hot encodings:
+
+
A useful thread has helped me to solve the downcasting issue in pd.DataFrame.replace() when trying to do one-hot encoding to replace the CYP strengths for each drug
+
A Pandas setting-with-copy warning shows if using df[“column_name”]:
+
+
+
A value is trying to be set on a copy of a slice from a DataFrame. Try using .loc[row_indexer,col_indexer] = value instead
+
+
The solution is to enable the copy-on-write globally (as commented in the code below; from Pandas reference).
+
+
PyTorch user warning appers if using df_train[“cyp_strength_of_evidence”].values, as this leads to non-writable tensors with a warning like this:
+
+
+
UserWarning: The given NumPy array is not writable, and PyTorch does not support non-writable tensors. This means writing to this tensor will result in undefined behavior. You may want to copy the array to protect its data or make it writable before converting it to a tensor. This type of warning will be suppressed for the rest of this program. (Triggered internally at /Users/runner/work/pytorch/pytorch/pytorch/torch/csrc/utils/tensor_numpy.cpp:212.)
+
+
One of the solutions is to add copy() e.g. col_encoded = one_hot(torch.from_numpy(df[“column_name”].values.copy()) % total_numbers_in_column) or alternatively, convert column into numpy array first, then make the numpy array writeable (which is what I’ve used in the code below).
+
+
+Code
+
## X_train
+# 1. Convert "cyp_strength_of_evidence" column into one-hot encoding
+# Enable copy-on-write globally to remove the warning
+pd.options.mode.copy_on_write =True
+
+# Replace CYP strength as numbers
+with pd.option_context('future.no_silent_downcasting', True):
+ df_train["cyp_strength_of_evidence"] = df_train["cyp_strength_of_evidence"].replace({"strong": 1, "mod": 2}).infer_objects()
+ df_test["cyp_strength_of_evidence"] = df_test["cyp_strength_of_evidence"].replace({"strong": 1, "mod": 2}).infer_objects()
+
+# Get total number of CYP strengths in df
+total_cyp_str_train =len(set(df_train["cyp_strength_of_evidence"]))
+
+# Convert column into numpy array first, then make the numpy array writeable
+cyp_array_train = df_train["cyp_strength_of_evidence"].to_numpy()
+cyp_array_train.flags.writeable =True
+cyp_str_train_t = one_hot(torch.from_numpy(cyp_array_train) % total_cyp_str_train)
+cyp_str_train_t
+
+
+
Without going into too much details about vector embeddings (as there are a lot of useful learning materials about it online and in texts), here’s roughly how I understand embeddings while working on this post. Embeddings are real-valued dense vectors that are normally in multi-dimensional arrays and they can represent and catch the context of a word or sentence, the semantic similarity and especially the relation of each word with other words in a corpus of texts. They roughly form the basis of natural language processing and also contribute to how large language models are built… in a very simplified sense, but obviously this can get complex if we want the models to do more. Here, I’m trying something experimental so I’m going to convert each ADR for each drug into embeddings.
+
+
+Code
+
# 2. Convert "adverse_drug_reactions" column into embeddings
+## see separate scripts used previously e.g. words_tensors.py
+## or Tensors_for_adrs_interactive.py to show step-by-step conversions from words to tensors
+
+# Save all ADRs from common ADRs column as a list (joining every row of ADRs in place only)
+adr_str_train = df_train["adverse_drug_reactions"].tolist()
+# Join separate rows of strings into one complete string
+adr_string_train =",".join(adr_str_train)
+# Converting all ADRs into Torch tensors using words_tensors.py
+from words_tensors import words_tensors
+adr_train_t = words_tensors(adr_string_train)
+adr_train_t
+
+
+
When trying to convert the “fp” column into tensors, there is one coding issue I’ve found relating to the data split step earlier. Each time the notebook is re-run with the kernel refreshed, the data split will lead to different proportions of training and testing sets due to the “shuffle = True”, which subsequently leads to different training and testing set arrays. One of the ways to circumvent this is to turn off the shuffle but this is not ideal for model training. So an alternative way that I’ve tried is to use ndarray.size (which is the product of elements in ndarray.shape, equivalent to multiplying the numbers of rows and columns), and divide the row of the intended tensor shape by 2 as I’m trying to reshape training arrays so they’re all in 2 columns in order for torch.cat() to work later.
+
+
+Code
+
# 3. Convert "fp" column into tensors
+# Stack numpy arrays in fingerprint column
+fp_train_array = np.stack(df_train["fp"])
+# Convert numpy array data type from uint32 to int32
+fp_train_array = fp_train_array.astype("int32")
+# Create tensors from array
+fp_train_t = torch.from_numpy(fp_train_array)
+# Reshape tensors
+fp_train_t = torch.reshape(fp_train_t, (int(fp_train_array.size/2), 2))
+fp_train_t.shape # tensor.ndim to check tensor dimensions
+
+
+
+
adr_train_t.shape
+
+
+
cyp_str_train_t.shape
+
+
+
# Concatenate adr tensors, fingerprint tensors and cyp strength tensors as X_train
+X_train = torch.cat([adr_train_t, fp_train_t, cyp_str_train_t], 0).float()
+X_train
## y_train
+# Use drug_class column as target
+# Convert "drug_class" column into embeddings
+# total number of drug classes in df = 20 - len(set(df["drug_class"])) - using embeddings instead of one-hot
+dc_str_train = df_train["drug_class"].tolist()
+dc_string_train =",".join(dc_str_train)
+y_train = words_tensors(dc_string_train)
+y_train
Input preprocessing pipeline using PyTorch Dataset and DataLoader
+
There is a size-mismatch-between-tensors warning when I’m trying to use PyTorch’s TensorDataset(). I’ve found out that to use the data loader and tensor dataset, the first dimension of all tensors needs to be the same. Initially, they’re not, where X_train.shape = [24313, 2], y_train.shape = [1, 2]. Eventually I’ve settled on two ways that can help with this:
+
+
use tensor.unsqueeze(dim = 1) or
+
use tensor[None] which’ll insert a new dimension at the beginning, then it becomes: X_train.shape = [1, 24313, 2], y_train.shape = [1, 1, 2]
+
+
+
X_train[None].shape
+
+
+
X_train.shape
+
+
+
y_train[None].shape
+
+
+
y_train.shape
+
+
+
# Create a PyTorch dataset on training data set
+train_data = TensorDataset(X_train[None], y_train[None])
+# Sets a seed number to generate random numbers
+torch.manual_seed(1)
+batch_size =1
+
+# Create a dataset loader
+train_dl = DataLoader(train_data, batch_size, shuffle =True)
+
+
+
# Create another PyTorch dataset on testing data set
+test_data = TensorDataset(X_test[None], y_test[None])
+torch.manual_seed(1)
+batch_size =1
+test_dl = DataLoader(test_data, batch_size, shuffle=True)
+
+
+
+
+
Set up a simple DNN regression model
+
I’m only going to use a very simple two-layer deep neural network model to match the tiny dataset used here. There are many other types of neural network layers or bits and pieces that can be used to suit the goals and purposes of the dataset used. This reference link shows different types of neural network layers that can be used in PyTorch.
+
Below are some short notes regarding a neural network (NN) model:
+
+
goal of the model is to minimise loss function L(W) (where W = weight) to get the optimal model weights
+
matrix with W (for hidden layer) connects input to hidden layer; matrix with W (for outer layer) connects hidden to output layer
+
Input layer -> activation function of hidden layer -> hidden layer -> activation function of output layer -> output layer (a very-simplified flow diagram to show how the layers get connected to each other)
+
+
About backpropagation for loss function:
+
+
backpropagation is a computationally efficient way to calculate partial derivatives of loss function to update weights in multi-layer NNs
+
it’s based on calculus chain rule to compute derivatives of mathematical functions (automatic differentiation)
+
matrix-vector multiplications in backpropagation are computationally more efficient to calculate than matrix-matrix multiplications e.g. forward propagation
# note: this is a very simple two-layer NN model only
+
+# Set up hidden units between two connected layers - one layer with 6 hidden units and the other with 3 hidden units
+hidden_units = [6, 3]
+# Input size same as number of columns in X_train
+input_size = X_train.shape[1]
+# Initiate NN layers as a list
+all_layers = []
+
+## Specify how the input, hidden and output layers are going to be connected
+# For each hidden unit within the hidden units specified above:
+for h_unit in hidden_units:
+# specify sizes of input sample (input size = X_train col size) & output sample (hidden units) in each layer
+# https://pytorch.org/docs/stable/generated/torch.nn.Linear.html#torch.nn.Linear
+ layer = nn.Linear(input_size, h_unit)
+# add each layer
+ all_layers.append(layer)
+# add activation function (trying rectified linear unit) for next layer
+ all_layers.append(nn.ReLU())
+# for the next layer to be added, the input size will be the same size as the hidden unit
+ input_size = h_unit
+
+# Specify the last layer (where input_feature = hidden_units[-1] = 3)
+all_layers.append(nn.Linear(hidden_units[-1], 1))
+
+# Set up a container that'll connect all layers in the specified sequence in the model
+model = nn.Sequential(*all_layers)
+model
+
+
+
+
+
+
Train model
+
This part is mainly about defining the loss function when training the model with the training data, and optimising model by using a stochastic gradient descent. One key thing I’ve gathered from trying to learn about deep learning is that we’re aiming for global minima and not local minima (e.g. if learning rate is too small, this may end up with local minima; if learning rate is too large, it may end up over-estimating the global minima). I’ve also encountered the PyTorch padding method to make sure the input and target tensors are of the same size, otherwise the model will run into matrix broadcasting issue (which will likely influence the results). The training loss appears to have converged when the epoch runs reach 100 and/or after (note this may vary due to shuffle data sampling)… (I also think my data size is way too small to show a clear contrast in training loss convergence).
+
References for: nn.MSELoss() - measures mean squared error between X and y, and nn.functional.pad() - pads tensor (increase tensor size)
+
Obtaining training loss via model training:
+
+
+Code
+
# Set up loss function
+loss_f = nn.MSELoss()
+# Set up stochastic gradient descent optimiser to optimise model (minimise loss) during training
+# lr = learning rate - default: 0.049787 (1*e^-3)
+optim = torch.optim.SGD(model.parameters(), lr=0.005)
+# Set training epochs (epoch: each cycle of training or passing through the training set)
+num_epochs =200
+# Set the log output to show training loss - for every 20 epochs
+log_epochs =20
+torch.manual_seed(1)
+# Create empty lists to save training loss (for training and testing/validation sets)
+train_epoch_loss = []
+test_epoch_loss = []
+
+# Predict via training X_batch & obtain train loss via loss function from X_batch & y_batch
+for epoch inrange(num_epochs):
+ train_loss =0
+for X_batch, y_batch in train_dl:
+# Make predictions
+ predict = model(X_batch)[:, 0]
+# Make input tensors the same size as y_batch tensors
+ predict_pad = F.pad(predict[None], pad=(1, 0, 0, 0))
+# Calculate training loss
+ loss = loss_f(predict_pad, y_batch)
+# Calculate gradients (backpropagations)
+ loss.backward(retain_graph=True)
+# Update parameters using gradients
+ optim.step()
+# Reset gradients back to zero
+ optim.zero_grad()
+ train_loss += loss.item()
+
+if epoch % log_epochs ==0:
+print(f"Epoch {epoch} Loss {train_loss/len(train_dl):.4f}")
+
+ train_epoch_loss.append(train_loss)
+
+
+
Obtaining test or validation loss:
+
+
+Code
+
# Predict via testing X_batch & obtain test loss
+for epoch inrange(num_epochs):
+ test_loss =0
+for X_batch, y_batch in test_dl:
+# Make predictions
+ predict_test = model(X_batch)[:, 0]
+# Make input tensors the same size as y_batch tensors
+ predict_pad_test = F.pad(predict_test[None], pad=(1, 0, 0, 0))
+# Calculate training loss
+ loss = loss_f(predict_pad_test, y_batch)
+# Calculate gradients (backpropagations)
+ loss.backward(retain_graph=True)
+# Update parameters using gradients
+ optim.step()
+# Reset gradients back to zero
+ optim.zero_grad()
+ test_loss += loss.item()
+
+if epoch % log_epochs ==0:
+print(f"Epoch {epoch} Loss {test_loss/len(test_dl):.4f}")
+
+ test_epoch_loss.append(test_loss)
+
+
+
Showing train and test losses over training epochs in a plot:
At the moment, when this notebook is re-run on a refreshed kernel, this leads to a different train and test split each time, and also leading to a different train and test (validation) loss each time. There may be two types of scenarios shown in the plot above where:
+
+
test loss is higher than train loss (overfitting) - showing the model may be way too simplified and is likely under-trained
+
train loss is higher than test loss (underfitting) - showing that the model may not have been trained well, and is unable to learn the features in the training data and apply them to the test data
+
+
When there are actually more training data available with also other hyperparameters fine tuned, it may be possible to see another scenario where both test loss and train loss are very similar in trend, meaning the model is being trained well and able to generalise the training to the unseen data.
+
To mitigate overfitting:
+
+
firstly there should be more training data than what I’ve had here
+
use L1 or L2 regularisation to minimise model complexity by adding penalities to large weights
+
use early stopping during model training to stop training the model when test loss is becoming higher than the train loss
+
use torch.nn.Dropout() to randomly drop out some of the neurons to ensure the exisiting neurons will learn features without being too reliant on other neighbouring neurons in the network
+
I’ll try the early stopping or drop out method in future posts since current post is relatively long already…
+
+
To overcome underfitting:
+
+
increase training epochs
+
minimise regularisation
+
consider building a more complex or deeper neural network model
+
+
+
+
+
Evaluate model
+
I’m keeping this post simple so I’ve only used mean squared error (MSE) and mean absolute error (MAE) to evaluate the model which has made a prediction on the test set. The smaller the MSE, the less error the model has when making predictions. However this is not the only metric that will determine if a model is optimal for predictions, as I’ve also noticed that every time there’s a different train and test split, the MAE and MSE values will vary too, so it appears that some splits will generate smaller MSE and other splits will lead to larger MSE.
+
+
+Code
+
# torch.no_grad() - disable gradient calculations to reduce memory usage for inference (also like a decorator)
+with torch.no_grad():
+ predict_test = model(X_test.float())[:, 0]
+# Padding target tensor with set size of [(1, 2)] as input tensor size will vary
+# when notebook is re-run each time due to butina split with sample shuffling
+# so need to pad the target tensor accordingly
+ y_test_pad = F.pad(y_test, pad=(predict_test[None].shape[1] - y_test.shape[1], 0, 0, 0))
+ loss_new = loss_f(predict_test[None], y_test_pad)
+print(f"MSE for test set: {loss_new.item():.4f}")
+print(f"MAE for test set: {nn.L1Loss()(predict_test[None], y_test_pad).item():.4f}")
I haven’t done feature standardisation for X_train which is to centre X_train mean and divide by its standard deviation, code may be like this, X_train_normalised = (X_train - np.mean(X_train))/np.std(X_train) (if used on training data, need to apply this to testing data too)
+
Training features are certainly too small, however, the main goal of this very first post is to get an overall idea of how to construct a baseline deep neural network (DNN) regression model. There are lots of other things that can be done to the ADRs data e.g. adding more drug molecular features and properties. I have essentially only used the initial molecular fingerprints generated when doing the data split to add a bit of molecular aspect in the training dataset.
+
I haven’t taken into account the frequencies of words (e.g. same drug classes and same ADR terms across different drugs) in the training and testing data, however, the aim of this first piece of work is also not a semantic analysis in natural language processing so this might not be needed…
+
There may be other PyTorch functions that I do not yet know about that will deal with small datasets e.g. perhaps torch.sparse may be useful?… so this piece is certainly not the only way to do it, but one of the many ways to work with small data
+
+
+
+
+
Acknowledgements
+
I’m very thankful for the existence of these references or websites below which have helped me understand (or scratch a small surface of) deep learning and also solve the coding issues mentioned in this post:
Strength of evidence that the drug is metabolised by CYP3A4/5 (as quoted from above web link):
+
+
Strong Evidence: the enzyme is majorly responsible for drug metabolism.
+
Moderate Evidence: the enzyme plays a significant but not exclusive role in drug metabolism or the supporting literature is not extensive.
+
+
+
+
+
drug_class data sources
+
This information can be found in many national drug formularies, drug reference textbooks e.g. Martindale, American society of health-System pharmacists’ (ASHP) drug information (DI) monographs, PubChem, ChEMBL, FDA, Micromedex etc. or online drug resources such as Drugs.com. For the particular small dataset collected and used in the notebook, the following reference sources for ADRs also contain information on therapeutic drug classes.
using the health professional version for ADRs which usually contains ADR references from pharmaceutical manufacturers’ medicines information data sheets, ASHP DI monographs or journal paper references
+
+
2nd-line as separate data checks:
+
+
NZ formulary (nzf) - likely only available to NZ residents only; other national formularies should contain very similar drug information
drugs.com_uk_di - UK drug information section in Drugs.com (equivalent to pharmaceutical manufacturers’ medicines information data sheets)
+
+
two main types of occurrences/frequencies used:
+
^^ - common > 10%,
+
^ - less common 1% to 10%,
+
(not going to include other ones with lower incidences e.g. less common at 0.1% to 1%, rare for less than 0.1% etc.)
+
+
+
+
+
Exceptions or notes for ADRs
+
+
nausea and vomiting applies to many drugs so won’t be included (almost every drug will have these ADRs, they can be alleviated with electrolytes replacements and anti-nausea meds or other non-med options; rash on the other hand can sometimes be serious and life-threatening e.g. Stevens-Johnson syndrome)
+
similar or overlapping adverse effects will be removed to keep only one adverse effect for the same drug e.g. adverse skin reactions, rash, urticaria - rash and urticaria will be removed as allergic skin reactions encompass both symptoms
+
for ADR terms with similar meanings, e.g. pyrexia/fever - fever is used instead (only one will be used)
+
ADR mentioned in common ADR category and repeated in the less common one will have the ADR recorded in the higher incidence rate (at > 10%) only
+
some ADRs can be dose-related or formulations-related e.g. injection site irritations or allergic reactions caused by excipients/fillers (aim is to investigate the relationships between ADRs and drugs via computational tools e.g. any patterns between ADRs & drugs so dose/formulations-related ADRS will not be recorded here)
+
some postmarketing adverse effects are for different age populations e.g. paediatric patients of up to 12 years of age or elderly people - for now all of them are labelled as “(pm)” to denote postmarketing reports and are not differentiated in age groups
+
+
+
Notes for specific drugs
+
+
hydrocortisone (a moderate CYP3A4 substrate) has no reported ADR frequencies at all for its ADRs as they are entirely dependent on the dosage and duration of use (ADRs tend to be unnoticeable at appropriate low doses for short durations)
+
terfenadine (a strong CYP3A4 substrate) is actually withdrawn from the market in 1990s due to QT prolongations
+
lercanidipine (a moderate CYP3A4 substrate) has nil reported ADRs of more than 1% but has a few postmarketing reports recorded
+
telaprevir (a moderate CYP3A4 substrate) is usually administered within a combination therapy (e.g. along with peginterferon alfa and ribavirin)
+
quinine (a moderate CYP3A4 substrate) has all of its ADRs reported without frequencies. The most common ADRs are presented as a cluster of symptoms (known as cinchonism) and can occur during overdoses (usually very toxic) and also normal doses. These symptoms include “…tinnitus, hearing impairment, headache, nausea, vomiting, abdominal pain, diarrhoea, visual disturbances (including blindness), arrhythmias (which can have a very rapid onset), convulsions (which can be intractable), and rashes.” (as quoted from NZ formulary v150 - 01 Dec 2024)
+
ribociclib (a moderate CYP3A4 substrate) has a listed ADR of on-treatment deaths, which were found to be associated with patients also taking letrozole or fulvestrant at the same time and/or in patients with underlying malignancy
+
+
+
+
+
+
Abbreviations used
+
+
ws = withdrawal symptoms
+
ADH = antidiuretic hormone
+
pm = postmarketing reports
+
CNS = central nervous system
+
CFTR = cystic fibrosis transmembrane regulator
+
c_diff = Clostridioides/Clostridium difficile
+
ZE = Zollinger-Ellison
+
MTOR = mammalian target of rapamycin (protein kinase)
+
AST = aspartate transaminase/aminotransferase
+
ALT = alanine transaminase/aminotransferase
+
ALP = alkaline phosphatase
+
GGT = gamma-glutamyltransferase
+
RTI = respiratory tract infection
+
UTI = urinary tract infection
+
LDH = lactate dehydrogenase
+
dd = dose and duration-dependent
+
pm_HIV_pit = postmarketing reports for HIV protease inhibitor therapy
+
pm_hep_cyto = postmarketing reports in cancer patients where drug was taken with hepatotoxic/cytotoxic chemotherapy and antibiotics
+
+
+
+
+
+
+
+
+
+
+
+
+
+
\ No newline at end of file
diff --git a/posts/22_Simple_dnn_adrs/2_ADR_regressor.html b/posts/22_Simple_dnn_adrs/2_ADR_regressor.html
new file mode 100644
index 0000000..9f15ee1
--- /dev/null
+++ b/posts/22_Simple_dnn_adrs/2_ADR_regressor.html
@@ -0,0 +1,1736 @@
+
+
+
+
+
+
+
+
+
+
+
+Home - Building a simple deep learning model about adverse drug reactions
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
Building a simple deep learning model about adverse drug reactions
Some of the code blocks have been folded to keep the post length a bit more manageable - click on the code links to see full code (only applies to the HTML version, not the Jupyter notebook version).
+
+
+
Import libraries
+
+
+Code
+
import pandas as pd
+import torch
+import torch.nn as nn
+import torch.nn.functional as F
+from torch.nn.functional import one_hot
+from torch.utils.data import TensorDataset, DataLoader
+import numpy as np
+import datamol as dm
+import rdkit
+from rdkit import Chem
+from rdkit.Chem import rdFingerprintGenerator
+import useful_rdkit_utils as uru
+import sys
+from matplotlib import pyplot as plt
+print(f"Pandas version used: {pd.__version__}")
+print(f"PyTorch version used: {torch.__version__}")
+print(f"NumPy version used: {np.__version__}")
+print(f"RDKit version used: {rdkit.__version__}")
+print(f"Python version used: {sys.version}")
+
+
+
Pandas version used: 2.2.3
+PyTorch version used: 2.2.2
+NumPy version used: 1.26.4
+RDKit version used: 2024.09.4
+Python version used: 3.12.7 (v3.12.7:0b05ead877f, Sep 30 2024, 23:18:00) [Clang 13.0.0 (clang-1300.0.29.30)]
+
+
+
+
+
+
Import adverse drug reactions (ADRs) data
+
This is an extremely small set of data compiled manually (by me) via references stated in the dataframe. For details about what and how the data are collected, I’ve prepared a separate post as a data note (add post link) to explain key things about the data. It may not lead to a very significant result but it is done as an example of what an early or basic deep neural network (DNN) model may look like. Ideally there should be more training data and also more features added or used. I’ve hypothetically set the goal of this introductory piece to predict therapeutic drug classes from ADRs, molecular fingerprints and cytochrome P450 substrate strengths, but this won’t be achieved in this initial post (yet).
+
+
data = pd.read_csv("All_CYP3A4_substrates")
+print(data.shape)
+data.head(3)
+
+
(27, 8)
+
+
+
+
+
+
+
+
+
+
generic_drug_name
+
notes
+
cyp_strength_of_evidence
+
drug_class
+
adverse_drug_reactions
+
first_ref
+
second_ref
+
date_checked
+
+
+
+
+
0
+
carbamazepine
+
NaN
+
strong
+
antiepileptics
+
constipation^^, leucopenia^^, dizziness^^, som...
+
drugs.com
+
nzf
+
211024
+
+
+
1
+
eliglustat
+
NaN
+
strong
+
metabolic_agents
+
diarrhea^^, oropharyngeal_pain^^, arthralgia^^...
+
drugs.com
+
emc
+
151124
+
+
+
2
+
flibanserin
+
NaN
+
strong
+
CNS_agents
+
dizziness^^, somnolence^^, sedation^, fatigue^...
+
drugs.com
+
Drugs@FDA
+
161124
+
+
+
+
+
+
+
For drug with astericks marked in “notes” column, see data notes under “Exceptions for ADRs” section in 1_ADR_data.qmd (separate post).
+
I’m dropping some of the columns that are not going to be used later.
Before extracting data from ChEMBL, I’m getting a list of drug names in capital letters ready first which can be fed into chembl_downloader with my old cyp_drugs.py to retrieve the SMILES of these drugs.
+
+
+Code
+
string = df["generic_drug_name"].tolist()
+# Convert list of drugs into multiple strings of drug names
+drugs =f"'{"','".join(string)}'"
+# Convert from lower case to upper case
+for letter in drugs:
+if letter.islower():
+ drugs = drugs.replace(letter, letter.upper())
+print(drugs)
# Get SMILES for each drug (via copying-and-pasting the previous cell output - attempted various ways to feed the string
+# directly into cyp_drugs.py, current way seems to be the most straightforward one...)
+from cyp_drugs import chembl_drugs
+# Using ChEMBL version 34
+df_3a4 = chembl_drugs(
+'CARBAMAZEPINE','ELIGLUSTAT','FLIBANSERIN','IMATINIB','IBRUTINIB','NERATINIB','ESOMEPRAZOLE','OMEPRAZOLE','IVACAFTOR','NALOXEGOL','OXYCODONE','SIROLIMUS','TERFENADINE','DIAZEPAM','HYDROCORTISONE','LANSOPRAZOLE','PANTOPRAZOLE','LERCANIDIPINE','NALDEMEDINE','NELFINAVIR','TELAPREVIR','ONDANSETRON','QUININE','RIBOCICLIB','SUVOREXANT','TELITHROMYCIN','TEMSIROLIMUS',
+#file_name="All_cyp3a4_smiles"
+ )
+print(df_3a4.shape)
+df_3a4.head(3)
+
+## Note: latest ChEMBL version 35 (as from 1st Dec 2024) seems to be taking a long time to load (no output after ~7min),
+## both versions 33 & 34 are ok with outputs loading within a few secs
+
+
+
(27, 4)
+
+
+
+
+
+
+
+
+
+
chembl_id
+
pref_name
+
max_phase
+
canonical_smiles
+
+
+
+
+
0
+
CHEMBL108
+
CARBAMAZEPINE
+
4
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
+
+
1
+
CHEMBL12
+
DIAZEPAM
+
4
+
CN1C(=O)CN=C(c2ccccc2)c2cc(Cl)ccc21
+
+
+
2
+
CHEMBL2110588
+
ELIGLUSTAT
+
4
+
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...
+
+
+
+
+
+
+
+
+
+
Merge dataframes
+
Next, I’m renaming the drug name column and merging the two dataframes together where one contains the ADRs and the other one contains the SMILES. I’m also making sure all drug names are in upper case for both dataframes so they can merge properly.
Then I’m parsing the canonical SMILES through my old script to generate these small molecules as RDKit molecules and standardised SMILES, making sure these SMILES are parsable.
+
+
+Code
+
# Using my previous code to preprocess small mols
+# disable rdkit messages
+dm.disable_rdkit_log()
+
+# The following function code were adapted from datamol.io
+def preprocess(row):
+
+"""
+ Function to preprocess, fix, standardise, sanitise compounds
+ and then generate various molecular representations based on these molecules.
+ Can be utilised as df.apply(preprocess, axis=1).
+
+ :param smiles_column: SMILES column name (needs to be names as "canonical_smiles")
+ derived from ChEMBL database (or any other sources) via an input dataframe
+ :param mol: RDKit molecules
+ :return: preprocessed RDKit molecules, standardised SMILES, SELFIES,
+ InChI and InChI keys added as separate columns in the dataframe
+ """
+
+# smiles_column = strings object
+ smiles_column ="canonical_smiles"
+# Convert each compound into a RDKit molecule in the smiles column
+ mol = dm.to_mol(row[smiles_column], ordered=True)
+# Fix common errors in the molecules
+ mol = dm.fix_mol(mol)
+# Sanitise the molecules
+ mol = dm.sanitize_mol(mol, sanifix=True, charge_neutral=False)
+# Standardise the molecules
+ mol = dm.standardize_mol(
+ mol,
+# Switch on to disconnect metal ions
+ disconnect_metals=True,
+ normalize=True,
+ reionize=True,
+# Switch on "uncharge" to neutralise charges
+ uncharge=True,
+# Taking care of stereochemistries of compounds
+# Note: this uses the older approach of "AssignStereochemistry()" from RDKit
+# https://github.com/datamol-io/datamol/blob/main/datamol/mol.py#L488
+ stereo=True,
+ )
+
+# Adding following rows of different molecular representations
+ row["rdkit_mol"] = dm.to_mol(mol)
+ row["standard_smiles"] = dm.standardize_smiles(str(dm.to_smiles(mol)))
+#row["selfies"] = dm.to_selfies(mol)
+#row["inchi"] = dm.to_inchi(mol)
+#row["inchikey"] = dm.to_inchikey(mol)
+return row
+
+df_p3a4 = df.apply(preprocess, axis =1)
+print(df_p3a4.shape)
+df_p3a4.head(3)
+
+
+
(27, 9)
+
+
+
+
+
+
+
+
+
+
pref_name
+
cyp_strength_of_evidence
+
drug_class
+
adverse_drug_reactions
+
chembl_id
+
max_phase
+
canonical_smiles
+
rdkit_mol
+
standard_smiles
+
+
+
+
+
0
+
CARBAMAZEPINE
+
strong
+
antiepileptics
+
constipation^^, leucopenia^^, dizziness^^, som...
+
CHEMBL108
+
4
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2dee0>
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
+
+
1
+
ELIGLUSTAT
+
strong
+
metabolic_agents
+
diarrhea^^, oropharyngeal_pain^^, arthralgia^^...
+
CHEMBL2110588
+
4
+
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2dfc0>
+
CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...
+
+
+
2
+
FLIBANSERIN
+
strong
+
CNS_agents
+
dizziness^^, somnolence^^, sedation^, fatigue^...
+
CHEMBL231068
+
4
+
O=c1[nH]c2ccccc2n1CCN1CCN(c2cccc(C(F)(F)F)c2)CC1
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2e030>
+
O=c1[nH]c2ccccc2n1CCN1CCN(c2cccc(C(F)(F)F)c2)CC1
+
+
+
+
+
+
+
+
+
+
Split data
+
Random splits usually lead to overly optimistic models, where testing molecules are too similar to traininig molecules leading to many problems. This is further discussed in two other blog posts that I’ve found useful - post by Greg Landrum and post by Pat Walters.
+
Here I’m trying out Pat’s useful_rdkit_utils’ GroupKFoldShuffle code (code originated from this thread) to split data (Butina clustering/splits). To do this, it requires SMILES to generate molecular fingerprints which will be used in the training and testing sets (potentially for future posts and in real-life cases, more things can be done with the SMILES or other molecular representations for machine learning, but to keep this post easy-to-read, I’ll stick with only generating the Morgan fingerprints for now).
+
+
+Code
+
# Generate numpy arrays containing the fingerprints
+df_p3a4['fp'] = df_p3a4.rdkit_mol.apply(rdFingerprintGenerator.GetMorganGenerator().GetCountFingerprintAsNumPy)
+
+# Get Butina cluster labels
+df_p3a4["butina_cluster"] = uru.get_butina_clusters(df_p3a4.standard_smiles)
+
+# Set up a GroupKFoldShuffle object
+group_kfold_shuffle = uru.GroupKFoldShuffle(n_splits=5, shuffle=True)
+
+# Using cross-validation/doing data split
+## X = np.stack(df_s3a4.fp), y = df.adverse_drug_reactions, group labels = df_s3a4.butina_cluster
+for train, test in group_kfold_shuffle.split(np.stack(df_p3a4.fp), df.adverse_drug_reactions, df_p3a4.butina_cluster):
+print(len(train),len(test))
+
+
+
17 10
+23 4
+23 4
+23 4
+22 5
+
+
+
+
+
+
Locate training and testing sets after data split
+
While trying to figure out how to locate training and testing sets after the data split, I’ve gone into a mini rabbit hole myself (a self-confusing session but gladly it clears up when my thought process goes further…). For example, some of the ways I’ve planned to try: create a dictionary as {index: butina label} first - butina cluster labels vs. index e.g. df_s3a4[“butina_cluster”], or maybe can directly convert from NumPy array to tensor - will need to locate drugs via indices first to specify training and testing sets, e.g. torch_train = torch.from_numpy(train) or torch_test = torch.from_numpy(test). It is actually simpler than this, which is to use pd.DataFrame.iloc() as shown below.
# What df_p3a4 now looks like after data split - with "fp" and "butina_cluster" columns added
+df_p3a4.head(1)
+
+
+
+
+
+
+
+
+
pref_name
+
cyp_strength_of_evidence
+
drug_class
+
adverse_drug_reactions
+
chembl_id
+
max_phase
+
canonical_smiles
+
rdkit_mol
+
standard_smiles
+
fp
+
butina_cluster
+
+
+
+
+
0
+
CARBAMAZEPINE
+
strong
+
antiepileptics
+
constipation^^, leucopenia^^, dizziness^^, som...
+
CHEMBL108
+
4
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
<rdkit.Chem.rdchem.Mol object at 0x13ef2dee0>
+
NC(=O)N1c2ccccc2C=Cc2ccccc21
+
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ...
+
20
+
+
+
+
+
+
+
+
# Convert indices into list
+train_set = train.tolist()
+# Locate drugs and drug info via pd.DataFrame.iloc
+df_train = df_p3a4.iloc[train_set]
+print(df_train.shape)
+df_train.head(2)
Set up training and testing sets for X and y variables
+
This part involves converting X (features) and y (target) variables into either one-hot encodings or vector embeddings, since I’ll be dealing with categories/words/ADRs and not numbers, and also to split each X and y variables into training and testing sets. At the very beginning, I’ve thought about using scikit_learn’s train_test_split(), but then realised that I should not need to do this as it’s already been done in the previous step (obviously I’m confusing myself again…). Essentially, this step can be integrated with the one-hot encoding and vector embeddings part as shown below.
+
There are three coding issues that have triggered warning messages when I’m trying to figure out how to convert CYP strengths into one-hot encodings:
+
+
A useful thread has helped me to solve the downcasting issue in pd.DataFrame.replace() when trying to do one-hot encoding to replace the CYP strengths for each drug
+
A Pandas setting-with-copy warning shows if using df[“column_name”]:
+
+
+
A value is trying to be set on a copy of a slice from a DataFrame. Try using .loc[row_indexer,col_indexer] = value instead
+
+
The solution is to enable the copy-on-write globally (as commented in the code below; from Pandas reference).
+
+
PyTorch user warning appers if using df_train[“cyp_strength_of_evidence”].values, as this leads to non-writable tensors with a warning like this:
+
+
+
UserWarning: The given NumPy array is not writable, and PyTorch does not support non-writable tensors. This means writing to this tensor will result in undefined behavior. You may want to copy the array to protect its data or make it writable before converting it to a tensor. This type of warning will be suppressed for the rest of this program. (Triggered internally at /Users/runner/work/pytorch/pytorch/pytorch/torch/csrc/utils/tensor_numpy.cpp:212.)
+
+
One of the solutions is to add copy() e.g. col_encoded = one_hot(torch.from_numpy(df[“column_name”].values.copy()) % total_numbers_in_column) or alternatively, convert column into numpy array first, then make the numpy array writeable (which is what I’ve used in the code below).
+
+
+Code
+
## X_train
+# 1. Convert "cyp_strength_of_evidence" column into one-hot encoding
+# Enable copy-on-write globally to remove the warning
+pd.options.mode.copy_on_write =True
+
+# Replace CYP strength as numbers
+with pd.option_context('future.no_silent_downcasting', True):
+ df_train["cyp_strength_of_evidence"] = df_train["cyp_strength_of_evidence"].replace({"strong": 1, "mod": 2}).infer_objects()
+ df_test["cyp_strength_of_evidence"] = df_test["cyp_strength_of_evidence"].replace({"strong": 1, "mod": 2}).infer_objects()
+
+# Get total number of CYP strengths in df
+total_cyp_str_train =len(set(df_train["cyp_strength_of_evidence"]))
+
+# Convert column into numpy array first, then make the numpy array writeable
+cyp_array_train = df_train["cyp_strength_of_evidence"].to_numpy()
+cyp_array_train.flags.writeable =True
+cyp_str_train_t = one_hot(torch.from_numpy(cyp_array_train) % total_cyp_str_train)
+cyp_str_train_t
Without going into too much details about vector embeddings (as there are a lot of useful learning materials about it online and in texts), here’s roughly how I understand embeddings while working on this post. Embeddings are real-valued dense vectors that are normally in multi-dimensional arrays and they can represent and catch the context of a word or sentence, the semantic similarity and especially the relation of each word with other words in a corpus of texts. They roughly form the basis of natural language processing and also contribute to how large language models are built… in a very simplified sense, but obviously this can get complex if we want the models to do more. Here, I’m trying something experimental so I’m going to convert each ADR for each drug into embeddings.
+
+
+Code
+
# 2. Convert "adverse_drug_reactions" column into embeddings
+## see separate scripts used previously e.g. words_tensors.py
+## or Tensors_for_adrs_interactive.py to show step-by-step conversions from words to tensors
+
+# Save all ADRs from common ADRs column as a list (joining every row of ADRs in place only)
+adr_str_train = df_train["adverse_drug_reactions"].tolist()
+# Join separate rows of strings into one complete string
+adr_string_train =",".join(adr_str_train)
+# Converting all ADRs into Torch tensors using words_tensors.py
+from words_tensors import words_tensors
+adr_train_t = words_tensors(adr_string_train)
+adr_train_t
When trying to convert the “fp” column into tensors, there is one coding issue I’ve found relating to the data split step earlier. Each time the notebook is re-run with the kernel refreshed, the data split will lead to different proportions of training and testing sets due to the “shuffle = True”, which subsequently leads to different training and testing set arrays. One of the ways to circumvent this is to turn off the shuffle but this is not ideal for model training. So an alternative way that I’ve tried is to use ndarray.size (which is the product of elements in ndarray.shape, equivalent to multiplying the numbers of rows and columns), and divide the row of the intended tensor shape by 2 as I’m trying to reshape training arrays so they’re all in 2 columns in order for torch.cat() to work later.
+
+
+Code
+
# 3. Convert "fp" column into tensors
+# Stack numpy arrays in fingerprint column
+fp_train_array = np.stack(df_train["fp"])
+# Convert numpy array data type from uint32 to int32
+fp_train_array = fp_train_array.astype("int32")
+# Create tensors from array
+fp_train_t = torch.from_numpy(fp_train_array)
+# Reshape tensors
+fp_train_t = torch.reshape(fp_train_t, (int(fp_train_array.size/2), 2))
+fp_train_t.shape # tensor.ndim to check tensor dimensions
+
+
+
torch.Size([22528, 2])
+
+
+
+
adr_train_t.shape
+
+
torch.Size([674, 2])
+
+
+
+
cyp_str_train_t.shape
+
+
torch.Size([22, 2])
+
+
+
+
# Concatenate adr tensors, fingerprint tensors and cyp strength tensors as X_train
+X_train = torch.cat([adr_train_t, fp_train_t, cyp_str_train_t], 0).float()
+X_train
## y_train
+# Use drug_class column as target
+# Convert "drug_class" column into embeddings
+# total number of drug classes in df = 20 - len(set(df["drug_class"])) - using embeddings instead of one-hot
+dc_str_train = df_train["drug_class"].tolist()
+dc_string_train =",".join(dc_str_train)
+y_train = words_tensors(dc_string_train)
+y_train
Input preprocessing pipeline using PyTorch Dataset and DataLoader
+
There is a size-mismatch-between-tensors warning when I’m trying to use PyTorch’s TensorDataset(). I’ve found out that to use the data loader and tensor dataset, the first dimension of all tensors needs to be the same. Initially, they’re not, where X_train.shape = [24313, 2], y_train.shape = [1, 2]. Eventually I’ve settled on two ways that can help with this:
+
+
use tensor.unsqueeze(dim = 1) or
+
use tensor[None] which’ll insert a new dimension at the beginning, then it becomes: X_train.shape = [1, 24313, 2], y_train.shape = [1, 1, 2]
+
+
+
X_train[None].shape
+
+
torch.Size([1, 23224, 2])
+
+
+
+
X_train.shape
+
+
torch.Size([23224, 2])
+
+
+
+
y_train[None].shape
+
+
torch.Size([1, 1, 2])
+
+
+
+
y_train.shape
+
+
torch.Size([1, 2])
+
+
+
+
# Create a PyTorch dataset on training data set
+train_data = TensorDataset(X_train[None], y_train[None])
+# Sets a seed number to generate random numbers
+torch.manual_seed(1)
+batch_size =1
+
+# Create a dataset loader
+train_dl = DataLoader(train_data, batch_size, shuffle =True)
+
+
+
# Create another PyTorch dataset on testing data set
+test_data = TensorDataset(X_test[None], y_test[None])
+torch.manual_seed(1)
+batch_size =1
+test_dl = DataLoader(test_data, batch_size, shuffle=True)
+
+
+
+
+
Set up a simple DNN regression model
+
I’m only going to use a very simple two-layer DNN model to match the tiny dataset used here. There are many other types of neural network layers or bits and pieces that can be used to suit the goals and purposes of the dataset used. This reference link shows different types of neural network layers that can be used in PyTorch.
+
Below are some short notes regarding a neural network (NN) model:
+
+
goal of the model is to minimise loss function L(W) (where W = weight) to get the optimal model weights
+
matrix with W (for hidden layer) connects input to hidden layer; matrix with W (for outer layer) connects hidden to output layer
+
Input layer -> activation function of hidden layer -> hidden layer -> activation function of output layer -> output layer (a very-simplified flow diagram to show how the layers get connected to each other)
+
+
About backpropagation for loss function:
+
+
backpropagation is a computationally efficient way to calculate partial derivatives of loss function to update weights in multi-layer NNs
+
it’s based on calculus chain rule to compute derivatives of mathematical functions (automatic differentiation)
+
matrix-vector multiplications in backpropagation are computationally more efficient to calculate than matrix-matrix multiplications e.g. forward propagation
# note: this is a very simple two-layer NN model only
+
+# Set up hidden units between two connected layers - one layer with 6 hidden units and the other with 3 hidden units
+hidden_units = [6, 3]
+# Input size same as number of columns in X_train
+input_size = X_train.shape[1]
+# Initiate NN layers as a list
+all_layers = []
+
+## Specify how the input, hidden and output layers are going to be connected
+# For each hidden unit within the hidden units specified above:
+for h_unit in hidden_units:
+# specify sizes of input sample (input size = X_train col size) & output sample (hidden units) in each layer
+# https://pytorch.org/docs/stable/generated/torch.nn.Linear.html#torch.nn.Linear
+ layer = nn.Linear(input_size, h_unit)
+# add each layer
+ all_layers.append(layer)
+# add activation function (trying rectified linear unit) for next layer
+ all_layers.append(nn.ReLU())
+# for the next layer to be added, the input size will be the same size as the hidden unit
+ input_size = h_unit
+
+# Specify the last layer (where input_feature = hidden_units[-1] = 3)
+all_layers.append(nn.Linear(hidden_units[-1], 1))
+
+# Set up a container that'll connect all layers in the specified sequence in the model
+model = nn.Sequential(*all_layers)
+model
This part is mainly about defining the loss function when training the model with the training data, and optimising model by using a stochastic gradient descent. One key thing I’ve gathered from trying to learn about deep learning is that we’re aiming for global minima and not local minima (e.g. if learning rate is too small, this may end up with local minima; if learning rate is too large, it may end up over-estimating the global minima). I’ve also encountered the PyTorch padding method to make sure the input and target tensors are of the same size, otherwise the model will run into matrix broadcasting issue (which will likely influence the results). The training loss appears to have converged when the epoch runs reach 100 and/or after (note this may vary due to shuffle data sampling)… (I also think my data size is way too small to show a clear contrast in training loss convergence).
+
References for: nn.MSELoss() - measures mean squared error between X and y, and nn.functional.pad() - pads tensor (increase tensor size)
+
Obtaining training loss via model training:
+
+
+Code
+
# Set up loss function
+loss_f = nn.MSELoss()
+# Set up stochastic gradient descent optimiser to optimise model (minimise loss) during training
+# lr = learning rate - default: 0.049787 (1*e^-3)
+optim = torch.optim.SGD(model.parameters(), lr=0.005)
+# Set training epochs (epoch: each cycle of training or passing through the training set)
+num_epochs =200
+# Set the log output to show training loss - for every 20 epochs
+log_epochs =20
+torch.manual_seed(1)
+# Create empty lists to save training loss (for training and testing/validation sets)
+train_epoch_loss = []
+test_epoch_loss = []
+
+# Predict via training X_batch & obtain train loss via loss function from X_batch & y_batch
+for epoch inrange(num_epochs):
+ train_loss =0
+for X_batch, y_batch in train_dl:
+# Make predictions
+ predict = model(X_batch)[:, 0]
+# Make input tensors the same size as y_batch tensors
+ predict_pad = F.pad(predict[None], pad=(1, 0, 0, 0))
+# Calculate training loss
+ loss = loss_f(predict_pad, y_batch)
+# Calculate gradients (backpropagations)
+ loss.backward(retain_graph=True)
+# Update parameters using gradients
+ optim.step()
+# Reset gradients back to zero
+ optim.zero_grad()
+ train_loss += loss.item()
+
+if epoch % log_epochs ==0:
+print(f"Epoch {epoch} Loss {train_loss/len(train_dl):.4f}")
+
+ train_epoch_loss.append(train_loss)
+
+
+
Epoch 0 Loss 4.3253
+Epoch 20 Loss 3.5549
+Epoch 40 Loss 2.9739
+Epoch 60 Loss 2.4838
+Epoch 80 Loss 2.1047
+
+
+
Epoch 100 Loss 1.8545
+Epoch 120 Loss 1.7217
+Epoch 140 Loss 1.6662
+Epoch 160 Loss 1.6471
+Epoch 180 Loss 1.6415
+
+
+
Obtaining test or validation loss:
+
+
+Code
+
# Predict via testing X_batch & obtain test loss
+for epoch inrange(num_epochs):
+ test_loss =0
+for X_batch, y_batch in test_dl:
+# Make predictions
+ predict_test = model(X_batch)[:, 0]
+# Make input tensors the same size as y_batch tensors
+ predict_pad_test = F.pad(predict_test[None], pad=(1, 0, 0, 0))
+# Calculate training loss
+ loss = loss_f(predict_pad_test, y_batch)
+# Calculate gradients (backpropagations)
+ loss.backward(retain_graph=True)
+# Update parameters using gradients
+ optim.step()
+# Reset gradients back to zero
+ optim.zero_grad()
+ test_loss += loss.item()
+
+if epoch % log_epochs ==0:
+print(f"Epoch {epoch} Loss {test_loss/len(test_dl):.4f}")
+
+ test_epoch_loss.append(test_loss)
+
+
+
Epoch 0 Loss 0.4037
+Epoch 20 Loss 0.1767
+Epoch 40 Loss 0.0963
+Epoch 60 Loss 0.0615
+Epoch 80 Loss 0.0452
+Epoch 100 Loss 0.0373
+Epoch 120 Loss 0.0335
+
+
+
Epoch 140 Loss 0.0316
+Epoch 160 Loss 0.0306
+Epoch 180 Loss 0.0301
+
+
+
+
+
+
Evaluate model
+
Showing train and test losses over training epochs in a plot:
At the moment, when this notebook is re-run on a refreshed kernel, this leads to a different train and test split each time, and also leading to a different train and test (validation) loss each time. There may be two types of scenarios shown in the plot above where:
+
+
test loss is higher than train loss (overfitting) - showing the model may be way too simplified and is likely under-trained
+
train loss is higher than test loss (underfitting) - showing that the model may not have been trained well, and is unable to learn the features in the training data and apply them to the test data
+
+
When there are actually more training data available with also other hyperparameters fine tuned, it may be possible to see another scenario where both test loss and train loss are very similar in trend, meaning the model is being trained well and able to generalise the training to the unseen data.
+
To mitigate overfitting:
+
+
firstly there should be more training data than what I’ve had here
+
use L1 or L2 regularisation to minimise model complexity by adding penalities to large weights
+
use early stopping during model training to stop training the model when test loss is becoming higher than the train loss
+
use torch.nn.Dropout() to randomly drop out some of the neurons to ensure the exisiting neurons will learn features without being too reliant on other neighbouring neurons in the network
+
I’ll try the early stopping or drop out method in future posts since current post is relatively long already…
+
+
To overcome underfitting:
+
+
increase training epochs
+
minimise regularisation
+
consider building a more complex or deeper neural network model
+
+
I’m trying to keep this post simple so have only used mean squared error (MSE) and mean absolute error (MAE) to evaluate the model which has made a prediction on the test set. The smaller the MSE, the less error the model has when making predictions. However this is not the only metric that will determine if a model is optimal for predictions, as I’ve also noticed that every time there’s a different train and test split, the MAE and MSE values will vary too, so it appears that some splits will generate smaller MSE and other splits will lead to larger MSE.
+
+
+Code
+
# torch.no_grad() - disable gradient calculations to reduce memory usage for inference (also like a decorator)
+with torch.no_grad():
+ predict_test = model(X_test.float())[:, 0]
+# Padding target tensor with set size of [(1, 2)] as input tensor size will vary
+# when notebook is re-run each time due to butina split with sample shuffling
+# so need to pad the target tensor accordingly
+ y_test_pad = F.pad(y_test, pad=(predict_test[None].shape[1] - y_test.shape[1], 0, 0, 0))
+ loss_new = loss_f(predict_test[None], y_test_pad)
+print(f"MSE for test set: {loss_new.item():.4f}")
+print(f"MAE for test set: {nn.L1Loss()(predict_test[None], y_test_pad).item():.4f}")
+
+
+
MSE for test set: 0.6576
+MAE for test set: 0.8070
I haven’t done feature standardisation for X_train which is to centre X_train mean and divide by its standard deviation, code may be like this, X_train_normalised = (X_train - np.mean(X_train))/np.std(X_train) (if used on training data, need to apply this to testing data too)
+
Training features are certainly too small, however, the main goal of this very first post is to get an overall idea of how to construct a baseline DNN regression model. There are lots of other things that can be done to the ADRs data e.g. adding more drug molecular features and properties. I have essentially only used the initial molecular fingerprints generated when doing the data split to add a bit of molecular aspect in the training dataset.
+
I haven’t taken into account the frequencies of words (e.g. same drug classes and same ADR terms across different drugs) in the training and testing data, however, the aim of this first piece of work is also not a semantic analysis in natural language processing so this might not be needed…
+
There may be other PyTorch functions that I do not yet know about that will deal with small datasets e.g. perhaps torch.sparse may be useful?… so this piece is certainly not the only way to do it, but one of the many ways to work with small data
+
+
+
+
+
Acknowledgements
+
I’m very thankful for the existence of these references, websites and reviewer below which have helped me understand (or scratch a small surface of) deep learning and also solve the coding issues mentioned in this post:
+
+
+
+
+
\ No newline at end of file
diff --git a/posts/22_Simple_dnn_adrs/2_ADR_regressor_files/figure-html/cell-33-output-2.png b/posts/22_Simple_dnn_adrs/2_ADR_regressor_files/figure-html/cell-33-output-2.png
new file mode 100644
index 0000000..2ad634a
Binary files /dev/null and b/posts/22_Simple_dnn_adrs/2_ADR_regressor_files/figure-html/cell-33-output-2.png differ
diff --git a/search.json b/search.json
index 1d16dfc..c638d0f 100644
--- a/search.json
+++ b/search.json
@@ -34,6 +34,20 @@
"section": "",
"text": "Introduction\nThis time I’m trying to build a web application in the hope to contribute my two cents towards minimising the gap between computational and laboratory sides in a drug discovery (or chemistry-related work) setting. There are definitely many other useful web applications available out there for similar uses, but I guess each one has its own uniqueness and special features.\nFor this app, it is mostly aimed at lab chemists who do not use any computer programming code at all in their work, and would like to quickly view compounds in the lab while working, and also to be able to highlight compound substructures during discussions or brainstorming for ideas during compound synthesis. Importantly, this app can exist outside a Jupyter notebook environment with internet required to access the app.\nThis is also the first part prior to the next post which will showcase the actual app. This part mainly involves some data cleaning but not as a major focus for this post. This is not to say that data cleaning is not important, but instead they are fundamental to any work involving data in order to ensure reasonable data quality, which can then potentially influence decisions or results. I have also collapsed the code sections below to make the post easier to read (to access code used for each section, click on the “Code” links).\n\n\n\nCode and explanations\nIt was actually surprisingly simple for this first part when I did it - building the interactive table. I came across this on LinkedIn on a random day for a post about itables being integrated with Shiny for Python plus Quarto. It came at the right time because I was actually trying to build this app. I quickly thought about incorporating it with the rest of the app so that users could refer back to the data quickly while visualising compound images. The code and explanations on building an interactive table for dataframes in Pandas and Polars were provided below.\nTo install itables, visit here for instructions and also for other information about supported notebook editors.\n\n\nCode\n# Import dataframe libraries\nimport pandas as pd\nimport polars as pl\n\n# Import Datamol\nimport datamol as dm\n\n# Import itables\nfrom itables import init_notebook_mode, show\ninit_notebook_mode(all_interactive=True)\n\n\n# Option 1: Reading df_ai.csv as a pandas dataframe\n#df = pd.read_csv(\"df_ai.csv\")\n#df.head\n\n# Option 2: Reading df_ai.csv as a polars dataframe\ndf = pl.read_csv(\"df_ai.csv\")\n#df.head()\n\n\n# Below was the code I used in my last post to fix the missing SMILES for neomycin \n# - the version below was edited due to recent updates in Polars\n# Canonical SMILES for neomycin was extracted from PubChem \n# (https://pubchem.ncbi.nlm.nih.gov/compound/Neomycin)\n\ndf = df.with_columns([\n pl.when(pl.col(\"Smiles\").str.lengths() == 0)\n .then(pl.lit(\"C1C(C(C(C(C1N)OC2C(C(C(C(O2)CN)O)O)N)OC3C(C(C(O3)CO)OC4C(C(C(C(O4)CN)O)O)N)O)O)N\"))\n .otherwise(pl.col(\"Smiles\"))\n .keep_name()\n])\n\n#df.head()\n\n\n\n\n\n\n\n\n\n\n\n\n\n\n\nPolars dataframe library was designed without the index in mind (which is different to Pandas), therefore the itables library did not work on my specific polars dataframe which required an index column to show (note: all other Polars dataframes should work fine with itables without the index column!).\nHowever to show row counts in Polars dataframes, we could use with_row_count() that starts the index from 0, and this would show up in a Jupyter environment as usual. A small code example would be like this below.\n\n\nCode\n# Uncomment below to run\n#df = df.with_row_count()\n\n\nThen I converted the Polars dataframe into a Pandas one (this could be completely avoided if you started with Pandas actually).\n\n\nCode\ndf = df.to_pandas()\n\n\nThen I added Datamol’s “_preprocess” function to convert SMILES1 into other molecular representations such as standardised SMILES (pre-processed and cleaned SMILES), SELFIES2, InChI3, InChI keys - just to provide extra information for further uses if needed. The standardised SMILES generated here would then be used for generating the molecule images later (in part 2).\n\n\nCode\n# Pre-process molecules using _preprocess function - adapted from datamol.io\n\nsmiles_column = \"Smiles\"\n\ndm.disable_rdkit_log()\n\ndef _preprocess(row):\n mol = dm.to_mol(row[smiles_column], ordered=True)\n mol = dm.fix_mol(mol)\n mol = dm.sanitize_mol(mol, sanifix=True, charge_neutral=False)\n mol = dm.standardize_mol(\n mol,\n disconnect_metals=False,\n normalize=True,\n reionize=True,\n uncharge=False,\n stereo=True,\n )\n\n row[\"standard_smiles\"] = dm.standardize_smiles(dm.to_smiles(mol))\n row[\"selfies\"] = dm.to_selfies(mol)\n row[\"inchi\"] = dm.to_inchi(mol)\n row[\"inchikey\"] = dm.to_inchikey(mol)\n return row\n\n#Apply the _preprocess function to the prepared dataframe.\ndf = df.apply(_preprocess, axis = 1)\n#df.head()\n\n\nThe next step was to keep the index column of the Pandas dataframe as an actual column (there was a reason for this, mainly for the app).\n\n\nCode\n# Convert index of Pandas df into a column\ndf = df.reset_index()\n#df.head()\n\n\n\n\n\nInteractive data table\n\nAn interactive table of all the prescription-only antibiotics from ChEMBL is shown below\nScroll the table from left to right to see the SMILES, standardised SMILES, SELFIES, InChI, InChI keys for each compound\nUse the search box to enter compound names to search for antibiotics and move between different pages when needed\n\n\n\nCode\ndf\n\n\n\n\n\n\n \n \n index\n Name\n Smiles\n standard_smiles\n selfies\n inchi\n inchikey\n \n Loading... (need help?)\n\n\n\n\n\n\n# Saving cleaned df_ai.csv as a new .csv file (for app_image_x.py - script to build the web app)\n# df = pl.from_pandas(df)\n# df.write_csv(\"df_ai_cleaned.csv\", sep = \",\")\n\n\n\n\nOptions for app deployment\nSince I had a lot of fun deploying my previous app in Shinylive last time, I thought I might try the same this time - deploying the Molviz app as a Shinylive app in Quarto. However, it didn’t work as expected, with reason being that RDKit wasn’t written in pure Python (it was written in Python and C++), so there wasn’t a pure Python wheel file available in PyPi - this link may provide some answers relating to this. Essentially, packages or libraries used in the app will need to be compatible with Pyodide in order for the Shinylive app to work. So, the most likely option to deploy this app now would be in Shinyapps.io or HuggingFace as I read about it recently.\n\n\n\nNext post\nCode and explanations for the actual Molviz app will be detailed in the next post. To access full code and files used for now, please visit this repository link.\n\n\n\n\n\nFootnotes\n\n\nSimplified Molecular Input Line Entry Systems↩︎\nSELF-referencIng Embedded Strings↩︎\nInternational Chemical Identifier↩︎"
},
+ {
+ "objectID": "posts/22_Simple_dnn_adrs/2_ADR_regressor.html",
+ "href": "posts/22_Simple_dnn_adrs/2_ADR_regressor.html",
+ "title": "Building a simple deep learning model about adverse drug reactions",
+ "section": "",
+ "text": "The notebook from this repository uses a venv created by using uv with a kernel set up this way.\nSome of the code blocks have been folded to keep the post length a bit more manageable - click on the code links to see full code (only applies to the HTML version, not the Jupyter notebook version).\n\n\nImport libraries\n\n\nCode\nimport pandas as pd\nimport torch\nimport torch.nn as nn\nimport torch.nn.functional as F\nfrom torch.nn.functional import one_hot\nfrom torch.utils.data import TensorDataset, DataLoader\nimport numpy as np\nimport datamol as dm\nimport rdkit\nfrom rdkit import Chem\nfrom rdkit.Chem import rdFingerprintGenerator\nimport useful_rdkit_utils as uru\nimport sys\nfrom matplotlib import pyplot as plt\nprint(f\"Pandas version used: {pd.__version__}\")\nprint(f\"PyTorch version used: {torch.__version__}\")\nprint(f\"NumPy version used: {np.__version__}\")\nprint(f\"RDKit version used: {rdkit.__version__}\")\nprint(f\"Python version used: {sys.version}\")\n\n\nPandas version used: 2.2.3\nPyTorch version used: 2.2.2\nNumPy version used: 1.26.4\nRDKit version used: 2024.09.4\nPython version used: 3.12.7 (v3.12.7:0b05ead877f, Sep 30 2024, 23:18:00) [Clang 13.0.0 (clang-1300.0.29.30)]\n\n\n\n\n\nImport adverse drug reactions (ADRs) data\nThis is an extremely small set of data compiled manually (by me) via references stated in the dataframe. For details about what and how the data are collected, I’ve prepared a separate post as a data note (add post link) to explain key things about the data. It may not lead to a very significant result but it is done as an example of what an early or basic deep neural network (DNN) model may look like. Ideally there should be more training data and also more features added or used. I’ve hypothetically set the goal of this introductory piece to predict therapeutic drug classes from ADRs, molecular fingerprints and cytochrome P450 substrate strengths, but this won’t be achieved in this initial post (yet).\n\ndata = pd.read_csv(\"All_CYP3A4_substrates\")\nprint(data.shape)\ndata.head(3)\n\n(27, 8)\n\n\n\n\n\n\n \n \n \n generic_drug_name\n notes\n cyp_strength_of_evidence\n drug_class\n adverse_drug_reactions\n first_ref\n second_ref\n date_checked\n \n \n \n \n 0\n carbamazepine\n NaN\n strong\n antiepileptics\n constipation^^, leucopenia^^, dizziness^^, som...\n drugs.com\n nzf\n 211024\n \n \n 1\n eliglustat\n NaN\n strong\n metabolic_agents\n diarrhea^^, oropharyngeal_pain^^, arthralgia^^...\n drugs.com\n emc\n 151124\n \n \n 2\n flibanserin\n NaN\n strong\n CNS_agents\n dizziness^^, somnolence^^, sedation^, fatigue^...\n drugs.com\n Drugs@FDA\n 161124\n \n \n\n\n\n\nFor drug with astericks marked in “notes” column, see data notes under “Exceptions for ADRs” section in 1_ADR_data.qmd (separate post).\nI’m dropping some of the columns that are not going to be used later.\n\n\nCode\ndf = data.drop([\n \"notes\",\n \"first_ref\", \n \"second_ref\", \n \"date_checked\"\n ], axis=1)\ndf.head(3)\n\n\n\n\n\n\n \n \n \n generic_drug_name\n cyp_strength_of_evidence\n drug_class\n adverse_drug_reactions\n \n \n \n \n 0\n carbamazepine\n strong\n antiepileptics\n constipation^^, leucopenia^^, dizziness^^, som...\n \n \n 1\n eliglustat\n strong\n metabolic_agents\n diarrhea^^, oropharyngeal_pain^^, arthralgia^^...\n \n \n 2\n flibanserin\n strong\n CNS_agents\n dizziness^^, somnolence^^, sedation^, fatigue^...\n \n \n\n\n\n\n\n\n\nImport SMILES data from ChEMBL\nBefore extracting data from ChEMBL, I’m getting a list of drug names in capital letters ready first which can be fed into chembl_downloader with my old cyp_drugs.py to retrieve the SMILES of these drugs.\n\n\nCode\nstring = df[\"generic_drug_name\"].tolist()\n# Convert list of drugs into multiple strings of drug names\ndrugs = f\"'{\"','\".join(string)}'\"\n# Convert from lower case to upper case\nfor letter in drugs:\n if letter.islower():\n drugs = drugs.replace(letter, letter.upper())\nprint(drugs)\n\n\n'CARBAMAZEPINE','ELIGLUSTAT','FLIBANSERIN','IMATINIB','IBRUTINIB','NERATINIB','ESOMEPRAZOLE','OMEPRAZOLE','IVACAFTOR','NALOXEGOL','OXYCODONE','SIROLIMUS','TERFENADINE','DIAZEPAM','HYDROCORTISONE','LANSOPRAZOLE','PANTOPRAZOLE','LERCANIDIPINE','NALDEMEDINE','NELFINAVIR','TELAPREVIR','ONDANSETRON','QUININE','RIBOCICLIB','SUVOREXANT','TELITHROMYCIN','TEMSIROLIMUS'\n\n\n\n\nCode\n# Get SMILES for each drug (via copying-and-pasting the previous cell output - attempted various ways to feed the string\n# directly into cyp_drugs.py, current way seems to be the most straightforward one...)\nfrom cyp_drugs import chembl_drugs\n# Using ChEMBL version 34\ndf_3a4 = chembl_drugs(\n 'CARBAMAZEPINE','ELIGLUSTAT','FLIBANSERIN','IMATINIB','IBRUTINIB','NERATINIB','ESOMEPRAZOLE','OMEPRAZOLE','IVACAFTOR','NALOXEGOL','OXYCODONE','SIROLIMUS','TERFENADINE','DIAZEPAM','HYDROCORTISONE','LANSOPRAZOLE','PANTOPRAZOLE','LERCANIDIPINE','NALDEMEDINE','NELFINAVIR','TELAPREVIR','ONDANSETRON','QUININE','RIBOCICLIB','SUVOREXANT','TELITHROMYCIN','TEMSIROLIMUS', \n #file_name=\"All_cyp3a4_smiles\"\n )\nprint(df_3a4.shape)\ndf_3a4.head(3)\n\n## Note: latest ChEMBL version 35 (as from 1st Dec 2024) seems to be taking a long time to load (no output after ~7min), \n## both versions 33 & 34 are ok with outputs loading within a few secs\n\n\n(27, 4)\n\n\n\n\n\n\n \n \n \n chembl_id\n pref_name\n max_phase\n canonical_smiles\n \n \n \n \n 0\n CHEMBL108\n CARBAMAZEPINE\n 4\n NC(=O)N1c2ccccc2C=Cc2ccccc21\n \n \n 1\n CHEMBL12\n DIAZEPAM\n 4\n CN1C(=O)CN=C(c2ccccc2)c2cc(Cl)ccc21\n \n \n 2\n CHEMBL2110588\n ELIGLUSTAT\n 4\n CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...\n \n \n\n\n\n\n\n\n\nMerge dataframes\nNext, I’m renaming the drug name column and merging the two dataframes together where one contains the ADRs and the other one contains the SMILES. I’m also making sure all drug names are in upper case for both dataframes so they can merge properly.\n\n\nCode\n# Rename column & change lower to uppercase\ndf = df.rename(columns={\"generic_drug_name\": \"pref_name\"})\ndf[\"pref_name\"] = df[\"pref_name\"].str.upper()\n# Merge df & df_3a4 \ndf = df.merge(df_3a4, how=\"left\", on=\"pref_name\")\ndf.head(3)\n\n\n\n\n\n\n \n \n \n pref_name\n cyp_strength_of_evidence\n drug_class\n adverse_drug_reactions\n chembl_id\n max_phase\n canonical_smiles\n \n \n \n \n 0\n CARBAMAZEPINE\n strong\n antiepileptics\n constipation^^, leucopenia^^, dizziness^^, som...\n CHEMBL108\n 4\n NC(=O)N1c2ccccc2C=Cc2ccccc21\n \n \n 1\n ELIGLUSTAT\n strong\n metabolic_agents\n diarrhea^^, oropharyngeal_pain^^, arthralgia^^...\n CHEMBL2110588\n 4\n CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...\n \n \n 2\n FLIBANSERIN\n strong\n CNS_agents\n dizziness^^, somnolence^^, sedation^, fatigue^...\n CHEMBL231068\n 4\n O=c1[nH]c2ccccc2n1CCN1CCN(c2cccc(C(F)(F)F)c2)CC1\n \n \n\n\n\n\n\n\n\nParse SMILES\nThen I’m parsing the canonical SMILES through my old script to generate these small molecules as RDKit molecules and standardised SMILES, making sure these SMILES are parsable.\n\n\nCode\n# Using my previous code to preprocess small mols\n# disable rdkit messages\ndm.disable_rdkit_log()\n\n# The following function code were adapted from datamol.io\ndef preprocess(row):\n\n \"\"\"\n Function to preprocess, fix, standardise, sanitise compounds \n and then generate various molecular representations based on these molecules.\n Can be utilised as df.apply(preprocess, axis=1).\n\n :param smiles_column: SMILES column name (needs to be names as \"canonical_smiles\") \n derived from ChEMBL database (or any other sources) via an input dataframe\n :param mol: RDKit molecules\n :return: preprocessed RDKit molecules, standardised SMILES, SELFIES, \n InChI and InChI keys added as separate columns in the dataframe\n \"\"\"\n\n # smiles_column = strings object\n smiles_column = \"canonical_smiles\"\n # Convert each compound into a RDKit molecule in the smiles column\n mol = dm.to_mol(row[smiles_column], ordered=True)\n # Fix common errors in the molecules\n mol = dm.fix_mol(mol)\n # Sanitise the molecules \n mol = dm.sanitize_mol(mol, sanifix=True, charge_neutral=False)\n # Standardise the molecules\n mol = dm.standardize_mol(\n mol,\n # Switch on to disconnect metal ions\n disconnect_metals=True,\n normalize=True,\n reionize=True,\n # Switch on \"uncharge\" to neutralise charges\n uncharge=True,\n # Taking care of stereochemistries of compounds\n # Note: this uses the older approach of \"AssignStereochemistry()\" from RDKit\n # https://github.com/datamol-io/datamol/blob/main/datamol/mol.py#L488\n stereo=True,\n )\n\n # Adding following rows of different molecular representations \n row[\"rdkit_mol\"] = dm.to_mol(mol)\n row[\"standard_smiles\"] = dm.standardize_smiles(str(dm.to_smiles(mol)))\n #row[\"selfies\"] = dm.to_selfies(mol)\n #row[\"inchi\"] = dm.to_inchi(mol)\n #row[\"inchikey\"] = dm.to_inchikey(mol)\n return row\n\ndf_p3a4 = df.apply(preprocess, axis = 1)\nprint(df_p3a4.shape)\ndf_p3a4.head(3)\n\n\n(27, 9)\n\n\n\n\n\n\n \n \n \n pref_name\n cyp_strength_of_evidence\n drug_class\n adverse_drug_reactions\n chembl_id\n max_phase\n canonical_smiles\n rdkit_mol\n standard_smiles\n \n \n \n \n 0\n CARBAMAZEPINE\n strong\n antiepileptics\n constipation^^, leucopenia^^, dizziness^^, som...\n CHEMBL108\n 4\n NC(=O)N1c2ccccc2C=Cc2ccccc21\n \n NC(=O)N1c2ccccc2C=Cc2ccccc21\n \n \n 1\n ELIGLUSTAT\n strong\n metabolic_agents\n diarrhea^^, oropharyngeal_pain^^, arthralgia^^...\n CHEMBL2110588\n 4\n CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...\n \n CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...\n \n \n 2\n FLIBANSERIN\n strong\n CNS_agents\n dizziness^^, somnolence^^, sedation^, fatigue^...\n CHEMBL231068\n 4\n O=c1[nH]c2ccccc2n1CCN1CCN(c2cccc(C(F)(F)F)c2)CC1\n \n O=c1[nH]c2ccccc2n1CCN1CCN(c2cccc(C(F)(F)F)c2)CC1\n \n \n\n\n\n\n\n\n\nSplit data\nRandom splits usually lead to overly optimistic models, where testing molecules are too similar to traininig molecules leading to many problems. This is further discussed in two other blog posts that I’ve found useful - post by Greg Landrum and post by Pat Walters.\nHere I’m trying out Pat’s useful_rdkit_utils’ GroupKFoldShuffle code (code originated from this thread) to split data (Butina clustering/splits). To do this, it requires SMILES to generate molecular fingerprints which will be used in the training and testing sets (potentially for future posts and in real-life cases, more things can be done with the SMILES or other molecular representations for machine learning, but to keep this post easy-to-read, I’ll stick with only generating the Morgan fingerprints for now).\n\n\nCode\n# Generate numpy arrays containing the fingerprints \ndf_p3a4['fp'] = df_p3a4.rdkit_mol.apply(rdFingerprintGenerator.GetMorganGenerator().GetCountFingerprintAsNumPy)\n\n# Get Butina cluster labels\ndf_p3a4[\"butina_cluster\"] = uru.get_butina_clusters(df_p3a4.standard_smiles)\n\n# Set up a GroupKFoldShuffle object\ngroup_kfold_shuffle = uru.GroupKFoldShuffle(n_splits=5, shuffle=True)\n\n# Using cross-validation/doing data split\n## X = np.stack(df_s3a4.fp), y = df.adverse_drug_reactions, group labels = df_s3a4.butina_cluster\nfor train, test in group_kfold_shuffle.split(np.stack(df_p3a4.fp), df.adverse_drug_reactions, df_p3a4.butina_cluster):\n print(len(train),len(test))\n\n\n17 10\n23 4\n23 4\n23 4\n22 5\n\n\n\n\n\nLocate training and testing sets after data split\nWhile trying to figure out how to locate training and testing sets after the data split, I’ve gone into a mini rabbit hole myself (a self-confusing session but gladly it clears up when my thought process goes further…). For example, some of the ways I’ve planned to try: create a dictionary as {index: butina label} first - butina cluster labels vs. index e.g. df_s3a4[“butina_cluster”], or maybe can directly convert from NumPy array to tensor - will need to locate drugs via indices first to specify training and testing sets, e.g. torch_train = torch.from_numpy(train) or torch_test = torch.from_numpy(test). It is actually simpler than this, which is to use pd.DataFrame.iloc() as shown below.\n\n# Training set indices\ntrain\n\narray([ 0, 1, 2, 3, 4, 6, 7, 8, 9, 10, 12, 13, 14, 15, 16, 17, 18,\n 20, 21, 22, 23, 25])\n\n\n\n# What df_p3a4 now looks like after data split - with \"fp\" and \"butina_cluster\" columns added\ndf_p3a4.head(1)\n\n\n\n\n\n \n \n \n pref_name\n cyp_strength_of_evidence\n drug_class\n adverse_drug_reactions\n chembl_id\n max_phase\n canonical_smiles\n rdkit_mol\n standard_smiles\n fp\n butina_cluster\n \n \n \n \n 0\n CARBAMAZEPINE\n strong\n antiepileptics\n constipation^^, leucopenia^^, dizziness^^, som...\n CHEMBL108\n 4\n NC(=O)N1c2ccccc2C=Cc2ccccc21\n \n NC(=O)N1c2ccccc2C=Cc2ccccc21\n [0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ...\n 20\n \n \n\n\n\n\n\n# Convert indices into list\ntrain_set = train.tolist()\n# Locate drugs and drug info via pd.DataFrame.iloc\ndf_train = df_p3a4.iloc[train_set]\nprint(df_train.shape)\ndf_train.head(2)\n\n(22, 11)\n\n\n\n\n\n\n \n \n \n pref_name\n cyp_strength_of_evidence\n drug_class\n adverse_drug_reactions\n chembl_id\n max_phase\n canonical_smiles\n rdkit_mol\n standard_smiles\n fp\n butina_cluster\n \n \n \n \n 0\n CARBAMAZEPINE\n strong\n antiepileptics\n constipation^^, leucopenia^^, dizziness^^, som...\n CHEMBL108\n 4\n NC(=O)N1c2ccccc2C=Cc2ccccc21\n \n NC(=O)N1c2ccccc2C=Cc2ccccc21\n [0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ...\n 20\n \n \n 1\n ELIGLUSTAT\n strong\n metabolic_agents\n diarrhea^^, oropharyngeal_pain^^, arthralgia^^...\n CHEMBL2110588\n 4\n CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...\n \n CCCCCCCC(=O)N[C@H](CN1CCCC1)[C@H](O)c1ccc2c(c1...\n [0, 2, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ...\n 19\n \n \n\n\n\n\n\n# Testing set indices\ntest\n\narray([ 5, 11, 19, 24, 26])\n\n\n\ntest_set = test.tolist()\ndf_test = df_p3a4.iloc[test_set]\nprint(df_test.shape)\ndf_test.head(2)\n\n(5, 11)\n\n\n\n\n\n\n \n \n \n pref_name\n cyp_strength_of_evidence\n drug_class\n adverse_drug_reactions\n chembl_id\n max_phase\n canonical_smiles\n rdkit_mol\n standard_smiles\n fp\n butina_cluster\n \n \n \n \n 5\n NERATINIB\n strong\n tyrosine_kinase_inhibitor\n diarrhea^^, abdominal_pain^^, stomatitis^^, dy...\n CHEMBL180022\n 4\n CCOc1cc2ncc(C#N)c(Nc3ccc(OCc4ccccn4)c(Cl)c3)c2...\n \n CCOc1cc2ncc(C#N)c(Nc3ccc(OCc4ccccn4)c(Cl)c3)c2...\n [0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, ...\n 15\n \n \n 11\n SIROLIMUS\n strong\n immunosuppressant\n hypertriglyceridemia^^, hypercholesterolemia^^...\n CHEMBL413\n 4\n CO[C@H]1C[C@@H]2CC[C@@H](C)[C@@](O)(O2)C(=O)C(...\n \n CO[C@H]1C[C@@H]2CC[C@@H](C)[C@@](O)(O2)C(=O)C(...\n [0, 1, 0, 0, 0, 0, 1, 1, 0, 0, 0, 0, 0, 0, 0, ...\n 2\n \n \n\n\n\n\n\n\n\nSet up training and testing sets for X and y variables\nThis part involves converting X (features) and y (target) variables into either one-hot encodings or vector embeddings, since I’ll be dealing with categories/words/ADRs and not numbers, and also to split each X and y variables into training and testing sets. At the very beginning, I’ve thought about using scikit_learn’s train_test_split(), but then realised that I should not need to do this as it’s already been done in the previous step (obviously I’m confusing myself again…). Essentially, this step can be integrated with the one-hot encoding and vector embeddings part as shown below.\nThere are three coding issues that have triggered warning messages when I’m trying to figure out how to convert CYP strengths into one-hot encodings:\n\nA useful thread has helped me to solve the downcasting issue in pd.DataFrame.replace() when trying to do one-hot encoding to replace the CYP strengths for each drug\nA Pandas setting-with-copy warning shows if using df[“column_name”]:\n\n\nA value is trying to be set on a copy of a slice from a DataFrame. Try using .loc[row_indexer,col_indexer] = value instead\n\nThe solution is to enable the copy-on-write globally (as commented in the code below; from Pandas reference).\n\nPyTorch user warning appers if using df_train[“cyp_strength_of_evidence”].values, as this leads to non-writable tensors with a warning like this:\n\n\nUserWarning: The given NumPy array is not writable, and PyTorch does not support non-writable tensors. This means writing to this tensor will result in undefined behavior. You may want to copy the array to protect its data or make it writable before converting it to a tensor. This type of warning will be suppressed for the rest of this program. (Triggered internally at /Users/runner/work/pytorch/pytorch/pytorch/torch/csrc/utils/tensor_numpy.cpp:212.)\n\nOne of the solutions is to add copy() e.g. col_encoded = one_hot(torch.from_numpy(df[“column_name”].values.copy()) % total_numbers_in_column) or alternatively, convert column into numpy array first, then make the numpy array writeable (which is what I’ve used in the code below).\n\n\nCode\n## X_train\n# 1. Convert \"cyp_strength_of_evidence\" column into one-hot encoding\n# Enable copy-on-write globally to remove the warning\npd.options.mode.copy_on_write = True\n\n# Replace CYP strength as numbers\nwith pd.option_context('future.no_silent_downcasting', True):\n df_train[\"cyp_strength_of_evidence\"] = df_train[\"cyp_strength_of_evidence\"].replace({\"strong\": 1, \"mod\": 2}).infer_objects()\n df_test[\"cyp_strength_of_evidence\"] = df_test[\"cyp_strength_of_evidence\"].replace({\"strong\": 1, \"mod\": 2}).infer_objects()\n\n# Get total number of CYP strengths in df\ntotal_cyp_str_train = len(set(df_train[\"cyp_strength_of_evidence\"]))\n\n# Convert column into numpy array first, then make the numpy array writeable\ncyp_array_train = df_train[\"cyp_strength_of_evidence\"].to_numpy()\ncyp_array_train.flags.writeable = True\ncyp_str_train_t = one_hot(torch.from_numpy(cyp_array_train) % total_cyp_str_train)\ncyp_str_train_t\n\n\ntensor([[0, 1],\n [0, 1],\n [0, 1],\n [0, 1],\n [0, 1],\n [0, 1],\n [0, 1],\n [0, 1],\n [0, 1],\n [0, 1],\n [0, 1],\n [1, 0],\n [1, 0],\n [1, 0],\n [1, 0],\n [1, 0],\n [1, 0],\n [1, 0],\n [1, 0],\n [1, 0],\n [1, 0],\n [1, 0]])\n\n\nWithout going into too much details about vector embeddings (as there are a lot of useful learning materials about it online and in texts), here’s roughly how I understand embeddings while working on this post. Embeddings are real-valued dense vectors that are normally in multi-dimensional arrays and they can represent and catch the context of a word or sentence, the semantic similarity and especially the relation of each word with other words in a corpus of texts. They roughly form the basis of natural language processing and also contribute to how large language models are built… in a very simplified sense, but obviously this can get complex if we want the models to do more. Here, I’m trying something experimental so I’m going to convert each ADR for each drug into embeddings.\n\n\nCode\n# 2. Convert \"adverse_drug_reactions\" column into embeddings\n## see separate scripts used previously e.g. words_tensors.py \n## or Tensors_for_adrs_interactive.py to show step-by-step conversions from words to tensors\n\n# Save all ADRs from common ADRs column as a list (joining every row of ADRs in place only)\nadr_str_train = df_train[\"adverse_drug_reactions\"].tolist()\n# Join separate rows of strings into one complete string\nadr_string_train = \",\".join(adr_str_train)\n# Converting all ADRs into Torch tensors using words_tensors.py\nfrom words_tensors import words_tensors\nadr_train_t = words_tensors(adr_string_train)\nadr_train_t\n\n\ntensor([[-1.5256, -0.7502],\n [-0.6540, -1.6095],\n [-0.1002, -0.6092],\n ...,\n [ 1.4006, -0.7007],\n [ 0.3303, 1.6160],\n [-0.4700, -0.6566]], grad_fn=)\n\n\nWhen trying to convert the “fp” column into tensors, there is one coding issue I’ve found relating to the data split step earlier. Each time the notebook is re-run with the kernel refreshed, the data split will lead to different proportions of training and testing sets due to the “shuffle = True”, which subsequently leads to different training and testing set arrays. One of the ways to circumvent this is to turn off the shuffle but this is not ideal for model training. So an alternative way that I’ve tried is to use ndarray.size (which is the product of elements in ndarray.shape, equivalent to multiplying the numbers of rows and columns), and divide the row of the intended tensor shape by 2 as I’m trying to reshape training arrays so they’re all in 2 columns in order for torch.cat() to work later.\n\n\nCode\n# 3. Convert \"fp\" column into tensors\n# Stack numpy arrays in fingerprint column\nfp_train_array = np.stack(df_train[\"fp\"])\n# Convert numpy array data type from uint32 to int32\nfp_train_array = fp_train_array.astype(\"int32\")\n# Create tensors from array\nfp_train_t = torch.from_numpy(fp_train_array)\n# Reshape tensors\nfp_train_t = torch.reshape(fp_train_t, (int(fp_train_array.size/2), 2))\nfp_train_t.shape # tensor.ndim to check tensor dimensions\n\n\ntorch.Size([22528, 2])\n\n\n\nadr_train_t.shape\n\ntorch.Size([674, 2])\n\n\n\ncyp_str_train_t.shape\n\ntorch.Size([22, 2])\n\n\n\n# Concatenate adr tensors, fingerprint tensors and cyp strength tensors as X_train\nX_train = torch.cat([adr_train_t, fp_train_t, cyp_str_train_t], 0).float()\nX_train\n\ntensor([[-1.5256, -0.7502],\n [-0.6540, -1.6095],\n [-0.1002, -0.6092],\n ...,\n [ 1.0000, 0.0000],\n [ 1.0000, 0.0000],\n [ 1.0000, 0.0000]], grad_fn=)\n\n\nX_test is being set up similarly as shown below.\n\n\nCode\n## X_test\n# 1. Convert \"cyp_strength_of_evidence\" into one-hot encodings\ntotal_cyp_str_test = len(set(df_test[\"cyp_strength_of_evidence\"]))\narray_test = df_test[\"cyp_strength_of_evidence\"].to_numpy()\narray_test.flags.writeable = True\ncyp_str_test_t = one_hot(torch.from_numpy(array_test) % total_cyp_str_test)\n\n# 2. Convert \"adverse_drug_reactions\" column into embeddings\nadr_str_test = df_test[\"adverse_drug_reactions\"].tolist()\nadr_string_test = \",\".join(adr_str_test)\nadr_test_t = words_tensors(adr_string_test)\n\n# 3. Convert \"fp\" column into tensors\nfp_test_array = np.stack(df_test[\"fp\"])\nfp_test_array = fp_test_array.astype(\"int32\")\nfp_test_t = torch.from_numpy(fp_test_array)\nfp_test_t = torch.reshape(fp_test_t, (int(fp_test_array.size/2),2))\n\n# Concatenate adr tensors, drug class tensors and cyp strength tensors as X_test\nX_test = torch.cat([cyp_str_test_t, adr_test_t, fp_test_t], 0).float()\nX_test\n\n\ntensor([[0., 1.],\n [0., 1.],\n [1., 0.],\n ...,\n [0., 0.],\n [0., 0.],\n [0., 0.]], grad_fn=)\n\n\nThis is followed by setting up y_train.\n\n\nCode\n## y_train\n# Use drug_class column as target\n# Convert \"drug_class\" column into embeddings \n# total number of drug classes in df = 20 - len(set(df[\"drug_class\"])) - using embeddings instead of one-hot\ndc_str_train = df_train[\"drug_class\"].tolist()\ndc_string_train = \",\".join(dc_str_train)\ny_train = words_tensors(dc_string_train)\ny_train\n\n\ntensor([[-1.8107, -1.9912]], grad_fn=)\n\n\nLastly, y_test is being specified.\n\n\nCode\n## y_test\n# Convert \"drug_class\" column into embeddings \ndc_str_test = df_test[\"drug_class\"].tolist()\ndc_string_test = \",\".join(dc_str_test)\ny_test = words_tensors(dc_string_test)\ny_test\n\n\ntensor([[-0.2434, -0.5782]], grad_fn=)\n\n\n\n\n\nInput preprocessing pipeline using PyTorch Dataset and DataLoader\nThere is a size-mismatch-between-tensors warning when I’m trying to use PyTorch’s TensorDataset(). I’ve found out that to use the data loader and tensor dataset, the first dimension of all tensors needs to be the same. Initially, they’re not, where X_train.shape = [24313, 2], y_train.shape = [1, 2]. Eventually I’ve settled on two ways that can help with this:\n\nuse tensor.unsqueeze(dim = 1) or\nuse tensor[None] which’ll insert a new dimension at the beginning, then it becomes: X_train.shape = [1, 24313, 2], y_train.shape = [1, 1, 2]\n\n\nX_train[None].shape\n\ntorch.Size([1, 23224, 2])\n\n\n\nX_train.shape\n\ntorch.Size([23224, 2])\n\n\n\ny_train[None].shape\n\ntorch.Size([1, 1, 2])\n\n\n\ny_train.shape\n\ntorch.Size([1, 2])\n\n\n\n# Create a PyTorch dataset on training data set\ntrain_data = TensorDataset(X_train[None], y_train[None])\n# Sets a seed number to generate random numbers\ntorch.manual_seed(1)\nbatch_size = 1\n\n# Create a dataset loader\ntrain_dl = DataLoader(train_data, batch_size, shuffle = True)\n\n\n# Create another PyTorch dataset on testing data set\ntest_data = TensorDataset(X_test[None], y_test[None])\ntorch.manual_seed(1)\nbatch_size = 1\ntest_dl = DataLoader(test_data, batch_size, shuffle=True)\n\n\n\n\nSet up a simple DNN regression model\nI’m only going to use a very simple two-layer DNN model to match the tiny dataset used here. There are many other types of neural network layers or bits and pieces that can be used to suit the goals and purposes of the dataset used. This reference link shows different types of neural network layers that can be used in PyTorch.\nBelow are some short notes regarding a neural network (NN) model:\n\ngoal of the model is to minimise loss function L(W) (where W = weight) to get the optimal model weights\nmatrix with W (for hidden layer) connects input to hidden layer; matrix with W (for outer layer) connects hidden to output layer\nInput layer -> activation function of hidden layer -> hidden layer -> activation function of output layer -> output layer (a very-simplified flow diagram to show how the layers get connected to each other)\n\nAbout backpropagation for loss function:\n\nbackpropagation is a computationally efficient way to calculate partial derivatives of loss function to update weights in multi-layer NNs\nit’s based on calculus chain rule to compute derivatives of mathematical functions (automatic differentiation)\nmatrix-vector multiplications in backpropagation are computationally more efficient to calculate than matrix-matrix multiplications e.g. forward propagation\n\nNote: there are also other types of activation functions available to use in PyTorch.\n\n\nCode\n# note: this is a very simple two-layer NN model only\n\n# Set up hidden units between two connected layers - one layer with 6 hidden units and the other with 3 hidden units\nhidden_units = [6, 3]\n# Input size same as number of columns in X_train\ninput_size = X_train.shape[1]\n# Initiate NN layers as a list\nall_layers = []\n\n## Specify how the input, hidden and output layers are going to be connected\n# For each hidden unit within the hidden units specified above:\nfor h_unit in hidden_units:\n # specify sizes of input sample (input size = X_train col size) & output sample (hidden units) in each layer\n # https://pytorch.org/docs/stable/generated/torch.nn.Linear.html#torch.nn.Linear\n layer = nn.Linear(input_size, h_unit)\n # add each layer\n all_layers.append(layer)\n # add activation function (trying rectified linear unit) for next layer\n all_layers.append(nn.ReLU())\n # for the next layer to be added, the input size will be the same size as the hidden unit\n input_size = h_unit\n\n# Specify the last layer (where input_feature = hidden_units[-1] = 3)\nall_layers.append(nn.Linear(hidden_units[-1], 1))\n\n# Set up a container that'll connect all layers in the specified sequence in the model\nmodel = nn.Sequential(*all_layers)\nmodel\n\n\nSequential(\n (0): Linear(in_features=2, out_features=6, bias=True)\n (1): ReLU()\n (2): Linear(in_features=6, out_features=3, bias=True)\n (3): ReLU()\n (4): Linear(in_features=3, out_features=1, bias=True)\n)\n\n\n\n\n\nTrain model\nThis part is mainly about defining the loss function when training the model with the training data, and optimising model by using a stochastic gradient descent. One key thing I’ve gathered from trying to learn about deep learning is that we’re aiming for global minima and not local minima (e.g. if learning rate is too small, this may end up with local minima; if learning rate is too large, it may end up over-estimating the global minima). I’ve also encountered the PyTorch padding method to make sure the input and target tensors are of the same size, otherwise the model will run into matrix broadcasting issue (which will likely influence the results). The training loss appears to have converged when the epoch runs reach 100 and/or after (note this may vary due to shuffle data sampling)… (I also think my data size is way too small to show a clear contrast in training loss convergence).\nReferences for: nn.MSELoss() - measures mean squared error between X and y, and nn.functional.pad() - pads tensor (increase tensor size)\nObtaining training loss via model training:\n\n\nCode\n# Set up loss function\nloss_f = nn.MSELoss()\n# Set up stochastic gradient descent optimiser to optimise model (minimise loss) during training \n# lr = learning rate - default: 0.049787 (1*e^-3)\noptim = torch.optim.SGD(model.parameters(), lr=0.005)\n# Set training epochs (epoch: each cycle of training or passing through the training set)\nnum_epochs = 200\n# Set the log output to show training loss - for every 20 epochs\nlog_epochs = 20\ntorch.manual_seed(1)\n# Create empty lists to save training loss (for training and testing/validation sets)\ntrain_epoch_loss = []\ntest_epoch_loss = []\n\n# Predict via training X_batch & obtain train loss via loss function from X_batch & y_batch\nfor epoch in range(num_epochs):\n train_loss = 0\n for X_batch, y_batch in train_dl:\n # Make predictions\n predict = model(X_batch)[:, 0]\n # Make input tensors the same size as y_batch tensors\n predict_pad = F.pad(predict[None], pad=(1, 0, 0, 0))\n # Calculate training loss\n loss = loss_f(predict_pad, y_batch)\n # Calculate gradients (backpropagations)\n loss.backward(retain_graph=True)\n # Update parameters using gradients\n optim.step()\n # Reset gradients back to zero\n optim.zero_grad()\n train_loss += loss.item()\n \n if epoch % log_epochs == 0:\n print(f\"Epoch {epoch} Loss {train_loss/len(train_dl):.4f}\")\n\n train_epoch_loss.append(train_loss)\n\n\nEpoch 0 Loss 4.3253\nEpoch 20 Loss 3.5549\nEpoch 40 Loss 2.9739\nEpoch 60 Loss 2.4838\nEpoch 80 Loss 2.1047\n\n\nEpoch 100 Loss 1.8545\nEpoch 120 Loss 1.7217\nEpoch 140 Loss 1.6662\nEpoch 160 Loss 1.6471\nEpoch 180 Loss 1.6415\n\n\nObtaining test or validation loss:\n\n\nCode\n# Predict via testing X_batch & obtain test loss \nfor epoch in range(num_epochs):\n test_loss = 0\n for X_batch, y_batch in test_dl:\n # Make predictions\n predict_test = model(X_batch)[:, 0]\n # Make input tensors the same size as y_batch tensors\n predict_pad_test = F.pad(predict_test[None], pad=(1, 0, 0, 0))\n # Calculate training loss\n loss = loss_f(predict_pad_test, y_batch)\n # Calculate gradients (backpropagations)\n loss.backward(retain_graph=True)\n # Update parameters using gradients\n optim.step()\n # Reset gradients back to zero\n optim.zero_grad()\n test_loss += loss.item()\n \n if epoch % log_epochs == 0:\n print(f\"Epoch {epoch} Loss {test_loss/len(test_dl):.4f}\")\n\n test_epoch_loss.append(test_loss)\n\n\nEpoch 0 Loss 0.4037\nEpoch 20 Loss 0.1767\nEpoch 40 Loss 0.0963\nEpoch 60 Loss 0.0615\nEpoch 80 Loss 0.0452\nEpoch 100 Loss 0.0373\nEpoch 120 Loss 0.0335\n\n\nEpoch 140 Loss 0.0316\nEpoch 160 Loss 0.0306\nEpoch 180 Loss 0.0301\n\n\n\n\n\nEvaluate model\nShowing train and test losses over training epochs in a plot:\n\n\nCode\nplt.plot(train_epoch_loss, label=\"train_loss\")\nplt.plot(test_epoch_loss, label=\"test_loss\")\nplt.xlabel(\"Epochs\")\nplt.ylabel(\"Loss\")\nplt.legend()\nplt.show\n\n\n\n\n\n\n\n\nAt the moment, when this notebook is re-run on a refreshed kernel, this leads to a different train and test split each time, and also leading to a different train and test (validation) loss each time. There may be two types of scenarios shown in the plot above where:\n\ntest loss is higher than train loss (overfitting) - showing the model may be way too simplified and is likely under-trained\ntrain loss is higher than test loss (underfitting) - showing that the model may not have been trained well, and is unable to learn the features in the training data and apply them to the test data\n\nWhen there are actually more training data available with also other hyperparameters fine tuned, it may be possible to see another scenario where both test loss and train loss are very similar in trend, meaning the model is being trained well and able to generalise the training to the unseen data.\nTo mitigate overfitting:\n\nfirstly there should be more training data than what I’ve had here\nuse L1 or L2 regularisation to minimise model complexity by adding penalities to large weights\nuse early stopping during model training to stop training the model when test loss is becoming higher than the train loss\nuse torch.nn.Dropout() to randomly drop out some of the neurons to ensure the exisiting neurons will learn features without being too reliant on other neighbouring neurons in the network\nI’ll try the early stopping or drop out method in future posts since current post is relatively long already…\n\nTo overcome underfitting:\n\nincrease training epochs\nminimise regularisation\nconsider building a more complex or deeper neural network model\n\nI’m trying to keep this post simple so have only used mean squared error (MSE) and mean absolute error (MAE) to evaluate the model which has made a prediction on the test set. The smaller the MSE, the less error the model has when making predictions. However this is not the only metric that will determine if a model is optimal for predictions, as I’ve also noticed that every time there’s a different train and test split, the MAE and MSE values will vary too, so it appears that some splits will generate smaller MSE and other splits will lead to larger MSE.\n\n\nCode\n# torch.no_grad() - disable gradient calculations to reduce memory usage for inference (also like a decorator)\nwith torch.no_grad():\n predict_test = model(X_test.float())[:, 0]\n # Padding target tensor with set size of [(1, 2)] as input tensor size will vary \n # when notebook is re-run each time due to butina split with sample shuffling\n # so need to pad the target tensor accordingly\n y_test_pad = F.pad(y_test, pad=(predict_test[None].shape[1] - y_test.shape[1], 0, 0, 0))\n loss_new = loss_f(predict_test[None], y_test_pad)\n print(f\"MSE for test set: {loss_new.item():.4f}\")\n print(f\"MAE for test set: {nn.L1Loss()(predict_test[None], y_test_pad).item():.4f}\")\n\n\nMSE for test set: 0.6576\nMAE for test set: 0.8070\n\n\n\n\n\nSave model\nOne way to save the model is like below.\n\npath = \"adr_regressor.pt\"\ntorch.save(model, path)\nmodel_reload = torch.load(path)\nmodel_reload.eval()\n\nSequential(\n (0): Linear(in_features=2, out_features=6, bias=True)\n (1): ReLU()\n (2): Linear(in_features=6, out_features=3, bias=True)\n (3): ReLU()\n (4): Linear(in_features=3, out_features=1, bias=True)\n)\n\n\n\n\n\nReload model\nThe saved model is reloaded below with a check to make sure the reloaded version is the same as the saved version.\nReferences for: torch.max and torch.argmax\n\npred_reload = model_reload(X_test)\ny_test_rel_pad = F.pad(y_test, pad=(pred_reload[None].shape[1] - y_test.shape[1], 0, 0, 0))\ncorrect = (torch.argmax(pred_reload, dim=1) == y_test_rel_pad).float()\naccuracy = correct.mean()\nprint(f\"Test accuracy: {accuracy:.4f}\")\n\nTest accuracy: 0.9996\n\n\n\nA few things to consider in the end:\n\nI haven’t done feature standardisation for X_train which is to centre X_train mean and divide by its standard deviation, code may be like this, X_train_normalised = (X_train - np.mean(X_train))/np.std(X_train) (if used on training data, need to apply this to testing data too)\nTraining features are certainly too small, however, the main goal of this very first post is to get an overall idea of how to construct a baseline DNN regression model. There are lots of other things that can be done to the ADRs data e.g. adding more drug molecular features and properties. I have essentially only used the initial molecular fingerprints generated when doing the data split to add a bit of molecular aspect in the training dataset.\nI haven’t taken into account the frequencies of words (e.g. same drug classes and same ADR terms across different drugs) in the training and testing data, however, the aim of this first piece of work is also not a semantic analysis in natural language processing so this might not be needed…\nThere may be other PyTorch functions that I do not yet know about that will deal with small datasets e.g. perhaps torch.sparse may be useful?… so this piece is certainly not the only way to do it, but one of the many ways to work with small data\n\n\n\n\nAcknowledgements\nI’m very thankful for the existence of these references, websites and reviewer below which have helped me understand (or scratch a small surface of) deep learning and also solve the coding issues mentioned in this post:\n\nPyTorch forums\nStack Overflow\nRaschka, Sebastian, Yuxi (Hayden) Liu, and Vahid Mirjalili. 2022. Machine Learning with PyTorch and Scikit-Learn. Birmingham, UK: Packt Publishing.\nNoel O’Boyle for feedback on this post"
+ },
+ {
+ "objectID": "posts/22_Simple_dnn_adrs/1_ADR_data.html",
+ "href": "posts/22_Simple_dnn_adrs/1_ADR_data.html",
+ "title": "Notes on adverse drug reactions (ADRs) data",
+ "section": "",
+ "text": "Here are the notes regarding the strong and moderate CYP3A4 substrates used in the data in the accompanying notebook.\n\n\ndrug_name and cyp_strength_of_evidence source\nThis is all based on the Flockhart table of drug interactions: https://drug-interactions.medicine.iu.edu/MainTable.aspx\nStrength of evidence that the drug is metabolised by CYP3A4/5 (as quoted from above web link):\n\nStrong Evidence: the enzyme is majorly responsible for drug metabolism.\nModerate Evidence: the enzyme plays a significant but not exclusive role in drug metabolism or the supporting literature is not extensive.\n\n\n\n\ndrug_class data sources\nThis information can be found in many national drug formularies, drug reference textbooks e.g. Martindale, American society of health-System pharmacists’ (ASHP) drug information (DI) monographs, PubChem, ChEMBL, FDA, Micromedex etc. or online drug resources such as Drugs.com. For the particular small dataset collected and used in the notebook, the following reference sources for ADRs also contain information on therapeutic drug classes.\n\n\n\nADRs data sources\n\n1st-line: Drugs.com\n\nusing the health professional version for ADRs which usually contains ADR references from pharmaceutical manufacturers’ medicines information data sheets, ASHP DI monographs or journal paper references\n\n2nd-line as separate data checks:\n\nNZ formulary (nzf) - likely only available to NZ residents only; other national formularies should contain very similar drug information\nelectronic medicines compendium (emc) - UK-based drug reference\nDrugs@FDA - US-based drug reference\ndrugs.com_uk_di - UK drug information section in Drugs.com (equivalent to pharmaceutical manufacturers’ medicines information data sheets)\n\ntwo main types of occurrences/frequencies used:\n^^ - common > 10%,\n^ - less common 1% to 10%,\n(not going to include other ones with lower incidences e.g. less common at 0.1% to 1%, rare for less than 0.1% etc.)\n\n\n\n\nExceptions or notes for ADRs\n\nnausea and vomiting applies to many drugs so won’t be included (almost every drug will have these ADRs, they can be alleviated with electrolytes replacements and anti-nausea meds or other non-med options; rash on the other hand can sometimes be serious and life-threatening e.g. Stevens-Johnson syndrome)\nsimilar or overlapping adverse effects will be removed to keep only one adverse effect for the same drug e.g. adverse skin reactions, rash, urticaria - rash and urticaria will be removed as allergic skin reactions encompass both symptoms\nfor ADR terms with similar meanings, e.g. pyrexia/fever - fever is used instead (only one will be used)\nADR mentioned in common ADR category and repeated in the less common one will have the ADR recorded in the higher incidence rate (at > 10%) only\nsome ADRs can be dose-related or formulations-related e.g. injection site irritations or allergic reactions caused by excipients/fillers (aim is to investigate the relationships between ADRs and drugs via computational tools e.g. any patterns between ADRs & drugs so dose/formulations-related ADRS will not be recorded here)\nsome postmarketing adverse effects are for different age populations e.g. paediatric patients of up to 12 years of age or elderly people - for now all of them are labelled as “(pm)” to denote postmarketing reports and are not differentiated in age groups\n\n\nNotes for specific drugs\n\nhydrocortisone (a moderate CYP3A4 substrate) has no reported ADR frequencies at all for its ADRs as they are entirely dependent on the dosage and duration of use (ADRs tend to be unnoticeable at appropriate low doses for short durations)\nterfenadine (a strong CYP3A4 substrate) is actually withdrawn from the market in 1990s due to QT prolongations\nlercanidipine (a moderate CYP3A4 substrate) has nil reported ADRs of more than 1% but has a few postmarketing reports recorded\ntelaprevir (a moderate CYP3A4 substrate) is usually administered within a combination therapy (e.g. along with peginterferon alfa and ribavirin)\nquinine (a moderate CYP3A4 substrate) has all of its ADRs reported without frequencies. The most common ADRs are presented as a cluster of symptoms (known as cinchonism) and can occur during overdoses (usually very toxic) and also normal doses. These symptoms include “…tinnitus, hearing impairment, headache, nausea, vomiting, abdominal pain, diarrhoea, visual disturbances (including blindness), arrhythmias (which can have a very rapid onset), convulsions (which can be intractable), and rashes.” (as quoted from NZ formulary v150 - 01 Dec 2024)\nribociclib (a moderate CYP3A4 substrate) has a listed ADR of on-treatment deaths, which were found to be associated with patients also taking letrozole or fulvestrant at the same time and/or in patients with underlying malignancy\n\n\n\n\n\nAbbreviations used\n\nws = withdrawal symptoms\nADH = antidiuretic hormone\npm = postmarketing reports\nCNS = central nervous system\nCFTR = cystic fibrosis transmembrane regulator\nc_diff = Clostridioides/Clostridium difficile\nZE = Zollinger-Ellison\nMTOR = mammalian target of rapamycin (protein kinase)\nAST = aspartate transaminase/aminotransferase\nALT = alanine transaminase/aminotransferase\nALP = alkaline phosphatase\nGGT = gamma-glutamyltransferase\nRTI = respiratory tract infection\nUTI = urinary tract infection\nLDH = lactate dehydrogenase\ndd = dose and duration-dependent\npm_HIV_pit = postmarketing reports for HIV protease inhibitor therapy\npm_hep_cyto = postmarketing reports in cancer patients where drug was taken with hepatotoxic/cytotoxic chemotherapy and antibiotics"
+ },
{
"objectID": "posts/02_Long_COVID_dashboard/Tableau_dashboard.html",
"href": "posts/02_Long_COVID_dashboard/Tableau_dashboard.html",
@@ -291,7 +305,7 @@
"href": "index.html",
"title": "Data in life",
"section": "",
- "text": "🌟Blog status🌟\nWelcome to my blog on selected cheminformatics, machine learning and data science projects in drug discovery.\n\nLatest project - Please see updated web links for logistic regression under machine learning below\nNext project in the pipeline - Likely a simple DNN model about ADRs first\n\n\n🌟Past projects🌟\nMachine learning\n\nTree series - Decision tree 1 - data collection and preprocessing, 2 - data preprocessing and transformation, 3 - model building and estimating experimental errors, Random forest - model building, imbalanced dataset, feature importances & hyperparameter tuning, Random forest classifier - more on imbalanced dataset, Boosted trees - AdaBoost, XGBoost and Scikit-mol\nLogistic regression 1 - Parquet file in Polars dataframe library, 2 - Preprocessing data in Polars dataframe library, 3 - Building logistic regression model using scikit-learn, 4 - Evaluating logistic regression model in scikit-learn, older long version\n\nData explorations\n\nCytochrome P450 and small drug molecules with a focus on CYP3A4 and CYP2D6 inhibitors\nWorking with scaffolds in small molecules - Manipulating SMILES strings\nMolecular similarities in selected COVID-19 antivirals - Using RDKit’s similarity map and fingerprint generator\n\nWeb applications\n\nMolecular visualisation web application - Interactive data table, Using Shiny for Python web application framework\nShinylive app in Python - Data preparation, Embedding app in Quarto document & using pyodide.http\n\n\n\n\n\n\n\n\n\n\n\n\n\nCytochrome P450 and small drug molecules\n\n\nCYP3A4 and 2D6 inhibitors\n\n\n\n\n\n\nAug 22, 2024\n\n\n27 min\n\n\n\n\n\n\n\n\nBoosted trees\n\n\nSeries 2.3.1 - AdaBoost, XGBoost and Scikit-mol\n\n\n\n\n\n\nJun 6, 2024\n\n\n17 min\n\n\n\n\n\n\n\n\nUsing Molstar in Quarto\n\n\n\n\n\n\n\n\n\nApr 6, 2024\n\n\n1 min\n\n\n\n\n\n\n\n\nRandom forest classifier\n\n\nSeries 2.2.1 - more on imbalanced dataset\n\n\n\n\n\n\nJan 17, 2024\n\n\n13 min\n\n\n\n\n\n\n\n\nRandom forest\n\n\nSeries 2.2 - model building, imbalanced dataset, feature importances & hyperparameter tuning\n\n\n\n\n\n\nNov 22, 2023\n\n\n24 min\n\n\n\n\n\n\n\n\nDecision tree\n\n\nSeries 2.1.1 - data collection and preprocessing\n\n\n\n\n\n\nSep 19, 2023\n\n\n12 min\n\n\n\n\n\n\n\n\nDecision tree\n\n\nSeries 2.1.2 - data preprocessing and transformation\n\n\n\n\n\n\nSep 19, 2023\n\n\n12 min\n\n\n\n\n\n\n\n\nDecision tree\n\n\nSeries 2.1.3 - model building and estimating experimental errors\n\n\n\n\n\n\nSep 19, 2023\n\n\n14 min\n\n\n\n\n\n\n\n\nMolecular visualisation (Molviz) web application\n\n\nUsing Shiny for Python web application framework - part 2\n\n\n\n\n\n\nAug 10, 2023\n\n\n12 min\n\n\n\n\n\n\n\n\nMolecular visualisation (Molviz) web application\n\n\nInteractive data table - part 1\n\n\n\n\n\n\nAug 10, 2023\n\n\n5 min\n\n\n\n\n\n\n\n\nWorking with scaffolds in small molecules\n\n\nManipulating SMILES strings\n\n\n\n\n\n\nJul 6, 2023\n\n\n19 min\n\n\n\n\n\n\n\n\nShinylive app in Python\n\n\nEmbedding app in Quarto document & using pyodide.http to import csv files\n\n\n\n\n\n\nMay 24, 2023\n\n\n8 min\n\n\n\n\n\n\n\n\nShinylive app in Python\n\n\nEmbedding app in Quarto document for compounds in COVID-19 clinical trials\n\n\n\n\n\n\nMay 8, 2023\n\n\n23 min\n\n\n\n\n\n\n\n\nShinylive app in Python\n\n\nData preparation for compounds in COVID-19 clinical trials\n\n\n\n\n\n\nMay 8, 2023\n\n\n11 min\n\n\n\n\n\n\n\n\nShiny app in R\n\n\nSmall molecules in ChEMBL database\n\n\n\n\n\n\nApr 7, 2023\n\n\n6 min\n\n\n\n\n\n\n\n\nPills dataset - Part 3\n\n\nUsing Rust for data visualisation\n\n\n\n\n\n\nFeb 14, 2023\n\n\n10 min\n\n\n\n\n\n\n\n\nPills dataset - Part 2\n\n\nText cleaning using Polars & visualising pills with Plotly\n\n\n\n\n\n\nJan 31, 2023\n\n\n13 min\n\n\n\n\n\n\n\n\nPills dataset - Part 1\n\n\nWeb scraping, Polars & Pandas dataframe libraries\n\n\n\n\n\n\nJan 21, 2023\n\n\n11 min\n\n\n\n\n\n\n\n\nSmall molecules in ChEMBL database\n\n\nSeries 1.1.1 - Parquet file in Polars dataframe library\n\n\n\n\n\n\nJan 4, 2023\n\n\n10 min\n\n\n\n\n\n\n\n\nSmall molecules in ChEMBL database\n\n\nSeries 1.1.3 - Building logistic regression model using scikit-learn\n\n\n\n\n\n\nJan 4, 2023\n\n\n6 min\n\n\n\n\n\n\n\n\nSmall molecules in ChEMBL database\n\n\nSeries 1.1.2 - Preprocessing data in Polars dataframe library\n\n\n\n\n\n\nJan 4, 2023\n\n\n10 min\n\n\n\n\n\n\n\n\nSmall molecules in ChEMBL database\n\n\nSeries 1.1.4 - Evaluating logistic regression model in scikit-learn\n\n\n\n\n\n\nJan 4, 2023\n\n\n10 min\n\n\n\n\n\n\n\n\nSmall molecules in ChEMBL database (old)\n\n\nSeries 1.1 - Polars dataframe library and machine learning in scikit-learn\n\n\n\n\n\n\nJan 4, 2023\n\n\n29 min\n\n\n\n\n\n\n\n\nMolecular similarities in selected COVID-19 antivirals\n\n\nUsing RDKit’s similarity map and fingerprint generator\n\n\n\n\n\n\nNov 19, 2022\n\n\n28 min\n\n\n\n\n\n\n\n\nPhD project\n\n\nA research saga that went through COVID\n\n\n\n\n\n\nOct 23, 2022\n\n\n6 min\n\n\n\n\n\n\n\n\nPublications\n\n\n\n\n\n\n\n\n\nOct 23, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nLong COVID - an update\n\n\nPDF table scraping, bar graph, interactive map & wordcloud\n\n\n\n\n\n\nSep 19, 2022\n\n\n9 min\n\n\n\n\n\n\n\n\nTable scraping from PDF\n\n\nUsing tabula-py in Python\n\n\n\n\n\n\nSep 15, 2022\n\n\n3 min\n\n\n\n\n\n\n\n\nBlog move\n\n\n\n\n\n\n\n\n\nAug 6, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nPhenotypes associated with rare diseases\n\n\n\n\n\n\n\n\n\nAug 2, 2022\n\n\n7 min\n\n\n\n\n\n\n\n\nEmbracing social network\n\n\n\n\n\n\n\n\n\nJul 15, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nNatural history of rare diseases - malformation syndrome\n\n\n\n\n\n\n\n\n\nJun 27, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nUpdate on portfolio\n\n\n\n\n\n\n\n\n\nJun 13, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nLong COVID data in SQL\n\n\n\n\n\n\n\n\n\nJun 5, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nPortfolio projects\n\n\n\n\n\n\n\n\n\nMay 31, 2022\n\n\n0 min\n\n\n\n\n\n\n\n\nLong COVID dashboard\n\n\n\n\n\n\n\n\n\nMay 31, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nDrugs in rare diseases\n\n\n\n\n\n\n\n\n\nMay 28, 2022\n\n\n3 min\n\n\n\n\n\n\n\n\nFocussing on data analytics\n\n\n\n\n\n\n\n\n\nApr 15, 2022\n\n\n0 min\n\n\n\n\n\n\n\n\nThe beginning of the data science journey\n\n\n\n\n\n\n\n\n\nJan 28, 2022\n\n\n3 min\n\n\n\n\n\n\n\n\nSERCA project\n\n\n\n\n\n\n\n\n\nJan 24, 2022\n\n\n2 min\n\n\n\n\n\n\nNo matching items"
+ "text": "🌟Blog status🌟\nWelcome to my blog on selected cheminformatics, machine learning and data science projects in drug discovery.\n\nLatest project - Please see updated web links for logistic regression under machine learning below\nNext project in the pipeline - Likely a simple DNN model about ADRs first\n\n\n🌟Past projects🌟\nMachine learning\n\nTree series - Decision tree 1 - data collection and preprocessing, 2 - data preprocessing and transformation, 3 - model building and estimating experimental errors, Random forest - model building, imbalanced dataset, feature importances & hyperparameter tuning, Random forest classifier - more on imbalanced dataset, Boosted trees - AdaBoost, XGBoost and Scikit-mol\nLogistic regression 1 - Parquet file in Polars dataframe library, 2 - Preprocessing data in Polars dataframe library, 3 - Building logistic regression model using scikit-learn, 4 - Evaluating logistic regression model in scikit-learn, older long version\n\nData explorations\n\nCytochrome P450 and small drug molecules with a focus on CYP3A4 and CYP2D6 inhibitors\nWorking with scaffolds in small molecules - Manipulating SMILES strings\nMolecular similarities in selected COVID-19 antivirals - Using RDKit’s similarity map and fingerprint generator\n\nWeb applications\n\nMolecular visualisation web application - Interactive data table, Using Shiny for Python web application framework\nShinylive app in Python - Data preparation, Embedding app in Quarto document & using pyodide.http\n\n\n\n\n\n\n\n\n\n\n\n\n\nBuilding a simple deep learning model about adverse drug reactions\n\n\n\n\n\n\n\n\n\nJan 8, 2025\n\n\n21 min\n\n\n\n\n\n\n\n\nNotes on adverse drug reactions (ADRs) data\n\n\nFor strong and moderate-strengths CYP3A4 substrates\n\n\n\n\n\n\nJan 8, 2025\n\n\n4 min\n\n\n\n\n\n\n\n\nCytochrome P450 and small drug molecules\n\n\nCYP3A4 and 2D6 inhibitors\n\n\n\n\n\n\nAug 22, 2024\n\n\n27 min\n\n\n\n\n\n\n\n\nBoosted trees\n\n\nSeries 2.3.1 - AdaBoost, XGBoost and Scikit-mol\n\n\n\n\n\n\nJun 6, 2024\n\n\n17 min\n\n\n\n\n\n\n\n\nUsing Molstar in Quarto\n\n\n\n\n\n\n\n\n\nApr 6, 2024\n\n\n1 min\n\n\n\n\n\n\n\n\nRandom forest classifier\n\n\nSeries 2.2.1 - more on imbalanced dataset\n\n\n\n\n\n\nJan 17, 2024\n\n\n13 min\n\n\n\n\n\n\n\n\nRandom forest\n\n\nSeries 2.2 - model building, imbalanced dataset, feature importances & hyperparameter tuning\n\n\n\n\n\n\nNov 22, 2023\n\n\n24 min\n\n\n\n\n\n\n\n\nDecision tree\n\n\nSeries 2.1.1 - data collection and preprocessing\n\n\n\n\n\n\nSep 19, 2023\n\n\n12 min\n\n\n\n\n\n\n\n\nDecision tree\n\n\nSeries 2.1.2 - data preprocessing and transformation\n\n\n\n\n\n\nSep 19, 2023\n\n\n12 min\n\n\n\n\n\n\n\n\nDecision tree\n\n\nSeries 2.1.3 - model building and estimating experimental errors\n\n\n\n\n\n\nSep 19, 2023\n\n\n14 min\n\n\n\n\n\n\n\n\nMolecular visualisation (Molviz) web application\n\n\nUsing Shiny for Python web application framework - part 2\n\n\n\n\n\n\nAug 10, 2023\n\n\n12 min\n\n\n\n\n\n\n\n\nMolecular visualisation (Molviz) web application\n\n\nInteractive data table - part 1\n\n\n\n\n\n\nAug 10, 2023\n\n\n5 min\n\n\n\n\n\n\n\n\nWorking with scaffolds in small molecules\n\n\nManipulating SMILES strings\n\n\n\n\n\n\nJul 6, 2023\n\n\n19 min\n\n\n\n\n\n\n\n\nShinylive app in Python\n\n\nEmbedding app in Quarto document & using pyodide.http to import csv files\n\n\n\n\n\n\nMay 24, 2023\n\n\n8 min\n\n\n\n\n\n\n\n\nShinylive app in Python\n\n\nEmbedding app in Quarto document for compounds in COVID-19 clinical trials\n\n\n\n\n\n\nMay 8, 2023\n\n\n23 min\n\n\n\n\n\n\n\n\nShinylive app in Python\n\n\nData preparation for compounds in COVID-19 clinical trials\n\n\n\n\n\n\nMay 8, 2023\n\n\n11 min\n\n\n\n\n\n\n\n\nShiny app in R\n\n\nSmall molecules in ChEMBL database\n\n\n\n\n\n\nApr 7, 2023\n\n\n6 min\n\n\n\n\n\n\n\n\nPills dataset - Part 3\n\n\nUsing Rust for data visualisation\n\n\n\n\n\n\nFeb 14, 2023\n\n\n10 min\n\n\n\n\n\n\n\n\nPills dataset - Part 2\n\n\nText cleaning using Polars & visualising pills with Plotly\n\n\n\n\n\n\nJan 31, 2023\n\n\n13 min\n\n\n\n\n\n\n\n\nPills dataset - Part 1\n\n\nWeb scraping, Polars & Pandas dataframe libraries\n\n\n\n\n\n\nJan 21, 2023\n\n\n11 min\n\n\n\n\n\n\n\n\nSmall molecules in ChEMBL database\n\n\nSeries 1.1.1 - Parquet file in Polars dataframe library\n\n\n\n\n\n\nJan 4, 2023\n\n\n10 min\n\n\n\n\n\n\n\n\nSmall molecules in ChEMBL database\n\n\nSeries 1.1.3 - Building logistic regression model using scikit-learn\n\n\n\n\n\n\nJan 4, 2023\n\n\n6 min\n\n\n\n\n\n\n\n\nSmall molecules in ChEMBL database\n\n\nSeries 1.1.2 - Preprocessing data in Polars dataframe library\n\n\n\n\n\n\nJan 4, 2023\n\n\n10 min\n\n\n\n\n\n\n\n\nSmall molecules in ChEMBL database\n\n\nSeries 1.1.4 - Evaluating logistic regression model in scikit-learn\n\n\n\n\n\n\nJan 4, 2023\n\n\n10 min\n\n\n\n\n\n\n\n\nSmall molecules in ChEMBL database (old)\n\n\nSeries 1.1 - Polars dataframe library and machine learning in scikit-learn\n\n\n\n\n\n\nJan 4, 2023\n\n\n29 min\n\n\n\n\n\n\n\n\nMolecular similarities in selected COVID-19 antivirals\n\n\nUsing RDKit’s similarity map and fingerprint generator\n\n\n\n\n\n\nNov 19, 2022\n\n\n28 min\n\n\n\n\n\n\n\n\nPhD project\n\n\nA research saga that went through COVID\n\n\n\n\n\n\nOct 23, 2022\n\n\n6 min\n\n\n\n\n\n\n\n\nPublications\n\n\n\n\n\n\n\n\n\nOct 23, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nLong COVID - an update\n\n\nPDF table scraping, bar graph, interactive map & wordcloud\n\n\n\n\n\n\nSep 19, 2022\n\n\n9 min\n\n\n\n\n\n\n\n\nTable scraping from PDF\n\n\nUsing tabula-py in Python\n\n\n\n\n\n\nSep 15, 2022\n\n\n3 min\n\n\n\n\n\n\n\n\nBlog move\n\n\n\n\n\n\n\n\n\nAug 6, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nPhenotypes associated with rare diseases\n\n\n\n\n\n\n\n\n\nAug 2, 2022\n\n\n7 min\n\n\n\n\n\n\n\n\nEmbracing social network\n\n\n\n\n\n\n\n\n\nJul 15, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nNatural history of rare diseases - malformation syndrome\n\n\n\n\n\n\n\n\n\nJun 27, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nUpdate on portfolio\n\n\n\n\n\n\n\n\n\nJun 13, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nLong COVID data in SQL\n\n\n\n\n\n\n\n\n\nJun 5, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nPortfolio projects\n\n\n\n\n\n\n\n\n\nMay 31, 2022\n\n\n0 min\n\n\n\n\n\n\n\n\nLong COVID dashboard\n\n\n\n\n\n\n\n\n\nMay 31, 2022\n\n\n1 min\n\n\n\n\n\n\n\n\nDrugs in rare diseases\n\n\n\n\n\n\n\n\n\nMay 28, 2022\n\n\n3 min\n\n\n\n\n\n\n\n\nFocussing on data analytics\n\n\n\n\n\n\n\n\n\nApr 15, 2022\n\n\n0 min\n\n\n\n\n\n\n\n\nThe beginning of the data science journey\n\n\n\n\n\n\n\n\n\nJan 28, 2022\n\n\n3 min\n\n\n\n\n\n\n\n\nSERCA project\n\n\n\n\n\n\n\n\n\nJan 24, 2022\n\n\n2 min\n\n\n\n\n\n\nNo matching items"
},
{
"objectID": "about.html",
diff --git a/sitemap.xml b/sitemap.xml
index 2958981..7d59e75 100644
--- a/sitemap.xml
+++ b/sitemap.xml
@@ -2,174 +2,182 @@
https://jhylin.github.io/Data_in_life_blog/posts/Blog-Portfolio_projects/Portfolio_projects.html
- 2024-12-22T07:48:58.385Z
+ 2025-01-08T07:34:03.368Zhttps://jhylin.github.io/Data_in_life_blog/posts/03_Long_COVID_data_in_SQL/Long_COVID_SQL.html
- 2024-12-22T07:48:59.677Z
+ 2025-01-08T07:34:04.567Zhttps://jhylin.github.io/Data_in_life_blog/posts/PhD_projects/PhD_projects.html
- 2024-12-22T07:49:01.002Z
+ 2025-01-08T07:34:05.889Zhttps://jhylin.github.io/Data_in_life_blog/posts/15_Molviz/Molviz.html
- 2024-12-22T07:49:02.636Z
+ 2025-01-08T07:34:07.525Zhttps://jhylin.github.io/Data_in_life_blog/posts/15_Molviz/itables.html
- 2024-12-22T07:49:04.346Z
+ 2025-01-08T07:34:09.232Z
+
+
+ https://jhylin.github.io/Data_in_life_blog/posts/22_Simple_dnn_adrs/2_ADR_regressor.html
+ 2025-01-08T07:34:11.308Z
+
+
+ https://jhylin.github.io/Data_in_life_blog/posts/22_Simple_dnn_adrs/1_ADR_data.html
+ 2025-01-08T07:34:13.058Zhttps://jhylin.github.io/Data_in_life_blog/posts/02_Long_COVID_dashboard/Tableau_dashboard.html
- 2024-12-22T07:49:05.618Z
+ 2025-01-08T07:34:14.309Zhttps://jhylin.github.io/Data_in_life_blog/posts/Blog-Data_analytics/Focussing_on_data_analytics.html
- 2024-12-22T07:49:06.734Z
+ 2025-01-08T07:34:15.749Zhttps://jhylin.github.io/Data_in_life_blog/posts/Blog-Social_network/Embracing_social_network.html
- 2024-12-22T07:49:07.975Z
+ 2025-01-08T07:34:17.117Zhttps://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_app_embed.html
- 2024-12-22T07:49:10.502Z
+ 2025-01-08T07:34:19.658Zhttps://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_app_embed_pyodide_http.html
- 2024-12-22T07:49:12.848Z
+ 2025-01-08T07:34:21.997Zhttps://jhylin.github.io/Data_in_life_blog/posts/13_Shiny_app_python/ShinyAppPy_PC_Cov19_data_prep.html
- 2024-12-22T07:49:16.203Z
+ 2025-01-08T07:34:26.428Zhttps://jhylin.github.io/Data_in_life_blog/posts/05_Phenotypes_associated_with_rare_diseases/Phenotypes_rare_diseases.html
- 2024-12-22T07:49:17.924Z
+ 2025-01-08T07:34:28.194Zhttps://jhylin.github.io/Data_in_life_blog/posts/18_Notes_molstar_quarto/Molstar_quarto.html
- 2024-12-22T07:49:20.246Z
+ 2025-01-08T07:34:30.390Zhttps://jhylin.github.io/Data_in_life_blog/posts/SERCA_project/SERCA_project.html
- 2024-12-22T07:49:22.197Z
+ 2025-01-08T07:34:32.435Zhttps://jhylin.github.io/Data_in_life_blog/posts/Publications/Side_projects.html
- 2024-12-22T07:49:22.550Z
+ 2025-01-08T07:34:32.863Zhttps://jhylin.github.io/Data_in_life_blog/posts/01_Drugs_in_rare_diseases/Rare_diseases_drugs.html
- 2024-12-22T07:49:23.898Z
+ 2025-01-08T07:34:34.052Zhttps://jhylin.github.io/Data_in_life_blog/posts/Blog-DS_journey/Beginning_of_DS_journey.html
- 2024-12-22T07:49:25.097Z
+ 2025-01-08T07:34:35.337Zhttps://jhylin.github.io/Data_in_life_blog/posts/Blog-Blog_move/Blog_move.html
- 2024-12-22T07:49:26.406Z
+ 2025-01-08T07:34:36.596Zhttps://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_evcxr_polars_plotly_final.html
- 2024-12-22T07:49:28.323Z
+ 2025-01-08T07:34:38.235Zhttps://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_polars_pills_df.html
- 2024-12-22T07:49:31.001Z
+ 2025-01-08T07:34:41.175Zhttps://jhylin.github.io/Data_in_life_blog/posts/09_Pills/Rust_polars_pills_ws.html
- 2024-12-22T07:49:34.786Z
+ 2025-01-08T07:34:45.139Zhttps://jhylin.github.io/Data_in_life_blog/posts/Blog-Update/Update_on_portfolio.html
- 2024-12-22T07:49:36.126Z
+ 2025-01-08T07:34:46.418Zhttps://jhylin.github.io/Data_in_life_blog/posts/21_ML1-1_Small_mols_in_chembl_update/ML1-1-1_chembl_cpds_parquet_new.html
- 2024-12-22T07:49:37.774Z
+ 2025-01-08T07:34:48.138Zhttps://jhylin.github.io/Data_in_life_blog/posts/21_ML1-1_Small_mols_in_chembl_update/ML1-1-3_chembl_cpds_ml_model.html
- 2024-12-22T07:49:39.452Z
+ 2025-01-08T07:34:49.771Zhttps://jhylin.github.io/Data_in_life_blog/posts/21_ML1-1_Small_mols_in_chembl_update/ML1-1-2_chembl_cpds_prep.html
- 2024-12-22T07:49:41.207Z
+ 2025-01-08T07:34:51.721Zhttps://jhylin.github.io/Data_in_life_blog/posts/21_ML1-1_Small_mols_in_chembl_update/ML1-1-4_chembl_cpds_evaluate.html
- 2024-12-22T07:49:42.930Z
+ 2025-01-08T07:34:53.467Zhttps://jhylin.github.io/Data_in_life_blog/posts/20_Cyp3a4_2d6_inh/1_CYP450_drugs.html
- 2024-12-22T07:49:47.226Z
+ 2025-01-08T07:34:57.812Zhttps://jhylin.github.io/Data_in_life_blog/posts/07_Molecular_similarities_in_COVID-19_antivirals/Mol_sim_covid.html
- 2024-12-22T07:49:49.943Z
+ 2025-01-08T07:35:00.969Zhttps://jhylin.github.io/Data_in_life_blog/posts/04_Natural_history_of_rare_diseases_–_malformation_syndrome/Natural_history_rare_diseases_mal_syn.html
- 2024-12-22T07:49:51.375Z
+ 2025-01-08T07:35:02.270Zhttps://jhylin.github.io/Data_in_life_blog/posts/19_ML2-3_Boosted_trees/1_adaboost_xgb.html
- 2024-12-22T07:49:54.101Z
+ 2025-01-08T07:35:05.179Zhttps://jhylin.github.io/Data_in_life_blog/posts/06_Long_COVID_update/ExtractTableFromPDF.html
- 2024-12-22T07:49:55.751Z
+ 2025-01-08T07:35:07.128Zhttps://jhylin.github.io/Data_in_life_blog/posts/06_Long_COVID_update/Long_COVID_update.html
- 2024-12-22T07:49:57.847Z
+ 2025-01-08T07:35:08.904Zhttps://jhylin.github.io/Data_in_life_blog/posts/14_Scaffolds_in_small_molecules/chembl_anti-inf_data_prep_current.html
- 2024-12-22T07:50:02.240Z
+ 2025-01-08T07:35:13.323Zhttps://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/1_data_col_prep.html
- 2024-12-22T07:50:04.585Z
+ 2025-01-08T07:35:15.444Zhttps://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/2_data_prep_tran.html
- 2024-12-22T07:50:06.900Z
+ 2025-01-08T07:35:17.854Zhttps://jhylin.github.io/Data_in_life_blog/posts/16_ML2-1_Decision_tree/3_model_build.html
- 2024-12-22T07:50:09.058Z
+ 2025-01-08T07:35:19.979Zhttps://jhylin.github.io/Data_in_life_blog/posts/08_ML1-1_Small_molecules_in_ChEMBL_database/ML1-1_chembl_cpds.html
- 2024-12-22T07:50:11.632Z
+ 2025-01-08T07:35:22.519Zhttps://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/2_random_forest_classifier.html
- 2024-12-22T07:50:14.229Z
+ 2025-01-08T07:35:25.032Zhttps://jhylin.github.io/Data_in_life_blog/posts/17_ML2-2_Random_forest/1_random_forest.html
- 2024-12-22T07:50:16.822Z
+ 2025-01-08T07:35:27.504Zhttps://jhylin.github.io/Data_in_life_blog/posts/12_Shiny_app_chembl/ShinyAppChembl.html
- 2024-12-22T07:50:18.074Z
+ 2025-01-08T07:35:28.956Zhttps://jhylin.github.io/Data_in_life_blog/ShinyAppChembl/Chembl_intro.html
- 2024-12-22T07:50:21.543Z
+ 2025-01-08T07:35:31.960Zhttps://jhylin.github.io/Data_in_life_blog/index.html
- 2024-12-22T07:50:22.512Z
+ 2025-01-08T07:35:32.919Zhttps://jhylin.github.io/Data_in_life_blog/about.html
- 2024-12-22T07:50:22.864Z
+ 2025-01-08T07:35:33.267Z