1. Upstream analysis
The project directory must be organized as follows:
/path/to/upstream/analysis/<user>/<project_name>
|-- raw
|-- sample1
|-- sample1_R1.fastq.gz
|-- sample2
|-- sample2_R1.fastq.gz
|-- config
|-- sample.<project_name>.pre.config
|-- system.<project_name>.pre.config
|-- amplicons.tsv
- Note that single-end read fastq files should be named with suffix
_R1.fastq.gz. -
<user>is the username of shinyApp
- Single/paired fastq files in .gz format for each sample
- Filled sample config file, example
- Filled system config file, example
- DNA methylation marker panel file, example
project="<project_name>"
sample_config="/path/to/upstream/analysis/<user>/${project}/config/sample.${project}.pre.config"
system_config="/path/to/upstream/analysis/<user>/${project}/config/system.${project}.pre.config"
The fastq files are located in /path/to/upstream/analysis/<user>/<project_name>/raw/
python "/path/to/pipe/run_Bpipe.py" $sample_config $system_config
/path/to/upstream/analysis/<user>/<project_name>
|-- raw
|-- sample1
|-- sample1_R1.fastq.gz
|-- config
|-- ref
|-- raw_trimmed
|-- sample1
|-- sample1_R1.fastq.gz
|-- sample1_R1.fastq.gz_trimming_report.txt
|-- sample1_R1_trimmed_fastqc.html
|-- aligned
|-- sample1
|-- sample1_R1.bam
|-- sample1_R1.bismark.bam
|-- called
|-- sample1
|-- sample1_R1.bismark.full.tsv
|-- sample1_R1.bismark.CpG_report.txt
|-- bigTable
|-- bigTable.tsv
|-- bigTable_filtered.tsv
|-- bigTable_filtered.tsv.coverage.tsv
|-- bigTable_filtered.tsv.methylation.csv
|-- bigTable_fullRead.tsv
|-- bigTable_fullRead_polymorphism.tsv
|-- bigTable_fullRead.tsv.coverage.tsv
|-- bigTable_fullRead.tsv.methylation.csv
|-- metrics
|-- metrics.tsv
|-- metrics_table.tsv
|-- trimmed_matrix.tsv
|-- fastqc_html.zip
|-- multiQC
|-- patterned
|-- sample1
|-- sample1.pattern
|-- frequency
|-- amplicon1.tsv
|-- amplicon2.tsv
|-- matrix
|-- amplicon1.tsv
|-- amplicon2.tsv
|-- pattern
|-- amplicon1.tsv
|-- amplicon1.pdf
|-- amplicon2.tsv
|-- amplicon2.pdf
|-- polymorphism
|-- amplicon1.polymorphism
|-- amplicon2.polymorphism
/srv/shiny-server/MethPanel
|-- app.R
|-- www
|-- full_read.png
Edit file /srv/shiny-server/MethPanel/app.R (file app.R on this GitHub) as follows:
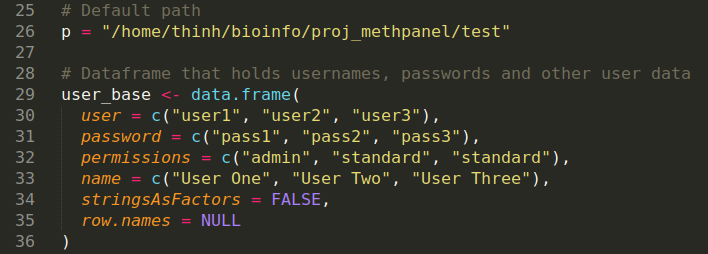
- Edit
/path/to/upstream/analysisto match your upstream analysis folder - Manually create password for each user, username must be the same as the
/path/to/upstream/analysis/<user>